

MaryO
-
Posts
8,063 -
Joined
-
Last visited
-
Days Won
548
Content Type
Profiles
Forums
Events
Blogs
Gallery
Articles
Media Demo
Store
Posts posted by MaryO
-
-
Boy, I really needed to reread this again today.
Many thanks again...to me!...for sharing.
Except for my new "hobby" of learning the balalaika, everything is seeming painful, blah, boring and, at times, plain annoying.
(this is my group, but was made a few years before I joined)
I have to try to remember that TODAY is The Best Day Of My Life - no matter what!

-
Abstract
In Cushing syndrome (CS), prolonged exposure to high cortisol levels results in a wide range of devastating effects causing multisystem morbidity. Despite the efficacy of treatment leading to disease remission and clinical improvement, hypercortisolism-induced complications may persist. Since glucocorticoids use the epigenetic machinery as a mechanism of action to modulate gene expression, the persistence of some comorbidities may be mediated by hypercortisolism-induced long-lasting epigenetic changes. Additionally, glucocorticoids influence microRNA expression, which is an important epigenetic regulator as it modulates gene expression without changing the DNA sequence. Evidence suggests that chronically elevated glucocorticoid levels may induce aberrant microRNA expression which may impact several cellular processes resulting in cardiometabolic disorders.
The present article reviews the evidence on epigenetic changes induced by (long-term) glucocorticoid exposure. Key aspects of some glucocorticoid-target genes and their implications in the context of CS are described. Lastly, the effects of epigenetic drugs influencing glucocorticoid effects are discussed for their ability to be potentially used as adjunctive therapy in CS.
Issue Section:Mini-reviewIn Cushing syndrome (CS), adrenocorticotropic hormone (ACTH) hypersecretion by a pituitary adenoma or an ectopic source, or autonomous cortisol hypersecretion by an adrenal tumor, induces chronic endogenous hypercortisolism with loss of the cortisol circadian rhythm (1). CS is more prevalent in women than men and frequently occurs in the fourth to sixth decades of life (2).
Glucocorticoids (GC) have extensive physiological actions and regulate up to 20% of the expressed genome, mainly related to the immune system, metabolic homeostasis, and cognition. Therefore, the prolonged exposure to high cortisol levels results in a wide range of devastating effects, including major changes in body composition (obesity, muscle atrophy, osteoporosis), neuropsychiatric disturbances (impaired cognition, depression, sleep disturbances), the metabolic syndrome (obesity, hypertension, insulin resistance, and dyslipidemia), hypercoagulability, and immune suppression (3, 4). The consequences of hypercortisolism lead to compromised quality of life and increased mortality rate (5). The mortality rate in patients with CS is 4 times higher than the healthy control population (6). Risk factors such as obesity, diabetes, and hypertension contribute to the increased risk of myocardial infarction, stroke, and cardiac insufficiency. As a result, cardiovascular disease is the leading cause of the premature death in CS (5). Infectious disease is also an important cause of death in CS (5). Therefore, prompt treatment to control hypercortisolism is imperative to prevent complications and an increased mortality rate.
Despite the efficacy of treatment leading to disease remission, the clinical burden of CS improves, but does not completely revert, in every patient (7). Indeed, obesity, neuropsychiatric disturbances, hypertension, diabetes, and osteoporosis persist in a substantial number of biochemically cured patients. For instance, in a study involving 118 CS patients in remission for about 7.8 years (median), resolution of comorbidities such as diabetes occurred in only 36% of cases, hypertension in 23% of cases, and depression in 52% of the cases (8). It has been proposed that epigenetic changes as a consequence of hypercortisolism is a mechanism of the persistence of some comorbidities (9-12).
Epigenetics is a reversible process that modifies gene expression without any alterations in DNA sequence; frequently it is mediated by histone modification and DNA methylation together with microRNAs (13-15). GCs use the epigenetic machinery as a mechanism of action to regulate gene expression in physiological circumstances, such as metabolic actions and stress response. Its networks involve DNA and histone modifying enzymes, such as DNA methyltransferases (DNMTs), histone acetyltransferases (HATs), and histone deacetylases (HDACs) (16). (Fig. 1) The DNA methylation process catalyzed by DNMTs is usually associated with downregulation of gene expression (17). Histone modifications catalyzed by HAT enzymes induce gene transcription, while those by HDAC enzymes induce transcriptional repression (17). Drugs interfering with these enzymes (so-called epigenetic drugs) may affect the GC genomic actions confirming the interaction between GC and the epigenetic system (18, 19). Furthermore, GC can modulate HDAC and DNMT expression and activity (16, 19, 20). Based on these data it might be speculated that in CS, epigenetic modifications induced by long-term GC exposure plays a role in the development of the disease-specific morbidity (9, 10).
Figure 1.Glucocorticoid (GC) and its epigenetic machinery. GC through its receptor interacts with DNA and histone modifying enzymes, such as DNA methyltransferases (DNMTs), histone acetyl transferases (HATs), and histone deacetylases (HDAC) to modulate gene expression.
In this review we provide an overview of epigenetic aspects of GC action in physiological conditions and in the context of CS. We start with a detailed characterization of how GC, using the epigenetic system, can change chromatin structure in order to activate or silence gene expression. (Fig. 2) Subsequently, we describe the role of epigenetic mechanisms in the regulation of expression of several GC-target genes related to CS. Finally, we present the current evidence of epigenetic changes caused by the long-term of GC exposure and the potential use of epidrugs influencing GC actions.
Figure 2.Epigenetic mechanisms of the glucocorticoid action to regulate gene expression. The GR is located in cytoplasm in a multi-protein complex; after GC binding, GR dissociates from the multi-protein complex, crosses the nuclear membrane, dimerizes, and binds to the GRE of the target gene. One of the mechanisms of action of GC is through the recruitment of co-regulators together with epigenetic enzymes, such as HAT, to change the chromatin structure, resulting in activation of gene transcription. Also, GR decreases gene expression by tethering other transcriptional factors and recruiting HDAC2, causing histone deacetylation, which leads to a repressed chromatin. GC can cause hypomethylation through downregulation in the expression of DNMT1. Abbreviations: Ac, acetylation; DNMT1, DNA methyltransferase 1; GC, glucocorticoid; GR, glucocorticoid receptor; GRE, glucocorticoid responsive elements; HAT, histone acetyltransferase; HDAC, histone deacetylases; Me: methylation.
Search Strategy
A search of the PubMed database was conducted using the advanced search builder tool for articles in the English language on the following terms “glucocorticoids,” “glucocorticoid receptor,” “Cushing,” “hypercortisolism,” “epigenetic,” “DNA methylation,” “histone deacetylase,” “histone acetyltransferase,” “microRNA” “fkbp5,” “clock genes,” and “POMC.” Moreover, references were identified directly from the articles included in this manuscript. The articles were selected by the authors after being carefully analyzed regarding their importance and impact.
Epigenetic Aspects of Genomic Action of Glucocorticoids
GCs regulate gene expression positively or negatively. GC-responsive genes include genes encoding for proteins associated with inflammation, metabolic processes, blood pressure and fluid homeostasis, apoptosis, cell cycle progression, circadian rhythm, and intracellular signaling (21).
The GC actions are cell type–specific (22). For instance, in an in vitro study, the comparison of GC-expressed genes between 2 cell lines, corticotroph (AtT20) and mammary (3134) cell lines, showed a different set of GC-regulated genes, revealing the cell type–specific nature of GC effects (23). GC function depends on the accessibility of glucocorticoid receptor (GR)-binding sites in the DNA of the target tissue, which in turn is mostly established during cell differentiation. Therefore, different chromatin organization explains the distinct GR-binding sites among different tissues (22, 24, 25). The chromatin accessibility is determined by histone modifications such as acetylation, methylation, phosphorylation, and/or DNA methylation, processes that are both dynamic and reversible (26).
Furthermore, gene expression is regulated in a GC-concentration-dependent manner which is tissue-specific. Only a few genes can be upregulated or downregulated at low concentrations of GC. For example, a dose of dexamethasone (Dex) as low as 0.5 nM selectively activated PER1 (period 1, transcription factor related to circadian rhythm) expression in lung cancer (A549) cells (21, 27). Additionally, continuous GC exposure or pulsed GC (cortisol fluctuation during circadian rhythm) may cause different responses with respect to gene expression (26, 28). For example, constant treatment with corticosterone induced higher levels of PER1 clock gene mRNA expression compared with pulsatile treatment, as demonstrated in an in vitro study using 3134 cell line (28).
The time course for gene expression in response to Dex is fast, with repression occurring slightly slower compared to activation. Half of activated and repressed genes are detected within, respectively, about 40 minutes and 53 minutes following Dex exposure (21).
In short, the transcriptional output in response to GC depends on cell type, as well as on the duration and intensity of GC exposure (21, 24, 26, 27). GCs act as a transcriptional regulatory factor resulting in activating or repressing the expression of genes. The GC exerts its function through binding to corticosteroid receptors, specifically, the mineralocorticoid receptor and the GR, members of the nuclear receptor superfamily (29, 30).
Glucocorticoid Receptor
The GR is located in the cytoplasm in a chaperone complex which includes heat-shock proteins (70 and 90) and immunophilins (such as FK506 binding protein [FKBP5]). Cortisol diffuses across the cell membrane and binds with high affinity to the GR. The activated GR bound to GC dissociates of the multi-protein complex and is transferred to the nucleus, where it ultimately regulates gene expression (26, 31).
GR is a transcription factor encoded by nuclear receptor subfamily 3, group C member 1 (NR3C1) gene, located in chromosome 5, and consisting of 9 exons. It is composed of 3 major functional domains, namely a DNA binding domain (DBD), the C-terminal ligand-binding domain (LBD) and the N-terminal domain (NTB). The LBD recognizes and joins the GC. NTB contains an activation function-1 (AF1) which connects with co-regulators and the members of the general transcription machinery to activate target genes. The DBD comprises 2 zinc fingers motifs that are able to identify and bind to glucocorticoid responsive elements (GREs) (32, 33).
GRα is the most expressed and functionally active GR. GRβ is another isoform which is the result of an alternative splicing in exon 9 of the GR transcript. The difference between the 2 isoforms is the distinct ligand-binding domain in GRβ. This variance prevents the GRβ from binding to GC. In fact, the GRβ counteracts GRα function by interfering with its binding to a GRE in the target gene, and GRβ expression is associated with GC resistance (32). In addition, GRβ has its own transcriptional activity which is independent and distinct from GRα (34).
Another splice variant of human GR, GRγ, is associated with GC resistance in lung cell carcinoma and childhood acute lymphoblastic leukemia (33, 35). There is an additional amino acid (arginine) in the DBD of the GRγ that reduces, by about half, the capacity to activate or suppress the transcription of the target gene, as compared with GRα (32). One study identified GRγ in a small series of corticotroph adenomas (36).
Glucocorticoid Mechanism of Action
The GR-GC complex induces or represses gene expression directly by binding to DNA, indirectly by tethering other transcription factors or yet in a composite manner that consists in binding DNA in association with binding to other co-regulators (35, 37).
The GR has the ability to reorganize the chromatin structure to become more or less accessible to the transcriptional machinery. In the classical mechanism of direct induction of gene expression, the GR dimerizes and binds to a GRE in DNA. The receptor recruits co-regulators, such as CREB binding protein, which has intrinsic histone acetyltransferase (HAT) activity that modifies the chromatin structure from an inactive to an active state. This model, called transactivation, upregulates the expression of some genes related to glucose, protein, and fat metabolism. Gene repression, on the other hand, is accomplished by GR binding to a negative GRE (nGRE) leading to the formation of a chromatin remodeling complex composed by co-repressor factors, such as NCOR1 and SMRT, and histone deacetylases (HDACs), that ultimately turn chromatin less accessible and suppress gene transcription. The gene repression through direct binding events occurs less frequently when compared to gene induction (25, 35, 38).
Another mechanism of GC action is through binding to other transcription factors (tethering). In case of switching off inflammatory genes, GR binds to transcriptional co-activator molecules, such as CREB binding protein with intrinsic HAT activity, and subsequently recruits HDAC2 to reverse histone acetylation, thus resulting in a suppression of the activated inflammatory gene (39). In the same model, GC interacts with other cofactors, such as the STAT family, to induce chromatin modifications resulting in increased gene expression (26).
Furthermore, the transcriptional dynamics of some genes follow a composite manner. In this model, GR, in conjunction with binding to GRE, also interacts with cofactors in order to enhance or reduce gene expression (35).
GCs can also modulate gene expression by influencing the transcription of epigenetic modifiers. An experimental study demonstrated that GC mediated the upregulation of HDAC2 in rats exposed to chronic stress, which in turn decreased the transcription of histone methyltransferase (Ehmt2) that ultimately upregulated the expression of Nedd4. Nedd4 is a ubiquitin ligase, expression of which has been related to cognitive impairment (40). Additionally, GC was found to interact with another epigenetic eraser, namely JMJD3, a histone demethylase, suppressing its transcription in endothelial cells treated with TNFα that led to decreased expression of other genes related to the blood-brain barrier (41).
GCs have the ability to induce (de)methylation changes in DNA, ultimately affecting gene expression. The DNA methylation process triggered by GC involves the family of DNA methyltransferases (DNMT) and ten-eleven translocation (TET) protein (20, 42-44). The DNMT, DNMT1, DNMT3A, and DNMT3B are able to transfer a methyl group to a cytosine residue in DNA, forming 5-methylcytosine (5mC), which negatively impacts gene expression. In contrast, TET protein chemically modifies the 5mC to form 5-hydroxymethylcytosine (5hmC), which ultimately leads to unmethylated cytosine, positively influencing gene expression (45).
Glucocorticoids mainly induce loss of methylation events rather than gain of methylation across the genome (11, 46). The DNA demethylation process can be either active or passive. The active mechanism is linked to the upregulation of TET enzyme expression that follows GC treatment, which was described in retinal and osteocyte cell line model studies (42, 43). The passive demethylation event involves the downregulation (Fig. 2) or dysfunction of DNMT1. DNMT1 is responsible for maintaining the methylation process in dividing cells (45). In case of GC exposure, GC can cause hypomethylation through downregulation in the expression of DNMT1, a process described in the AtT20 corticotroph tumor cell model, or through GC hindering DNMT activity, particularly DNMT1, as demonstrated in the retinal cell (RPE) line (20, 42, 44).
Glucocorticoid-Induced Epigenetic Changes
There are several molecular mechanisms connecting GR activation and epigenetic modifications ultimately affecting gene expression (Fig. 2). As described above, GC uses epigenetic machinery, such as DNA and histone modifying enzymes, to restructure the chromatin in order to induce or silence gene transcription (16, 47).
In an in vitro study using murine AtT20 corticotroph tumor and neuronal cell lines, after chronic GC exposure followed by a recovery period in the absence of GC, the cells retained an “epigenetic memory” with persistence of loss of methylation content in FKBP5 gene but with no increased gene expression at baseline. The functionality of this “epigenetic memory” only became evident in a second exposure to GC, when the cells responded sharply with a more robust expression of FKBP5 gene compared to the cells without previous exposure to GC (44). Another in vitro study, using a human fetal hippocampal cell line, confirmed long-lasting DNA methylation changes induced by GC. The cells were treated for 10 days with dexamethasone, during the proliferative and cell differentiation phases of the cell line, followed by 20 days without any treatment. The second exposure to GC resulted in an enhanced gene expression of a subset of GC-target genes (48). Additionally, using an animal model subjected to chronic stress, a distinct gene expression profile was demonstrated in response to acute GC challenge compared to those without chronic stress history. The proposed mechanism was that chronic stress resulted in GC-induced enduring epigenetic changes in target genes, altering the responsiveness to a subsequent GC exposure (49).
In general, it seems that the majority of differential methylation regions (DMRs) induced by GC are loss of methylation rather than gain of methylation. In an experimental study, an association between hypomethylation and GC exposure was demonstrated in mice previously exposed to high levels of GC. Further analysis demonstrated that the genes linked with DMR were mostly related to metabolism, the immune system, and neurodevelopment (11).
Human studies have also shown that excess of cortisol can induce modifications in DNA methylation. DNA methylation data obtained from whole blood samples from patients with chronic obstructive pulmonary disease (COPD) treated with GC revealed DMR at specific CpG dinucleotides across the genome. These DMR were confirmed by pyrosequencing and annotated to genes, such as SCNN1A, encoding the α subunit of the epithelial sodium channel, GPR97, encoding G protein coupled receptor 97, and LRP3, encoding low-density lipoprotein receptor-related protein 3 (50). Furthermore, it has been proposed that the negative impact of chronic GC exposure on the immune system, which increases the risk of opportunistically infections, may be epigenetically mediated (51). In a clinical study, using whole blood samples, an analysis of genome-wide DNA methylation was performed on patients before and after exposure to GC (51). Long-term GC exposure disrupts, through a persistent modification of the cytosine methylation pattern, the mTORC1 pathway which affects CD4+ T cell biology (51).
Taken together, these data clearly show the interplay between GC signaling and methylation and histone modifications processes suggesting that GC interferes in the epigenetic landscape modulating gene expression. It is possible that most of these GC-induced epigenetic events are dynamic and temporary, while others may persist leading to long-lasting disorders. Further research to provide insight into what makes some events reversible is warranted.
Epigenetic Changes as a Consequence of Long-Term Glucocorticoid Exposure in Cushing Syndrome
The comorbidities associated with CS are associated with increased mortality mainly due to cardiovascular events (52). GC-induced comorbidities in CS may be at least in part epigenetically mediated. Previous study using whole blood methylation profile demonstrated that specific hypomethylated CpG sites induced by GC were associated with Cushing comorbidities, such as hypertension and osteoporosis (46). The study identified a methylator predictor of GC excess which could be used as a biomarker to monitor GC status (46).
The long-term exposure to high cortisol levels may be crucial for the persistence of some morbidities in CS through epigenetic changes. Hypercortisolism-induced persistent changes in visceral adipose tissue gene expression through epigenetic modifications was investigated in a translational study (12). This study combined data from patients with active CS and data from an animal model of CS in active and remitted phase. Interestingly, the study demonstrated long-lasting changes in the transcriptome of adipose tissue that were associated with histone modifications induced by GC. Therefore, these epigenetic fingerprints observed even after the resolution of hypercortisolism may elucidate the mechanism of persistent modifications in gene expression in the visceral adipose tissue (12).
With regard to the persistence of GC-induced DMR, a genome-wide DNA methylation analysis showed a lower average of DNA methylation in patients in remission of CS compared to controls. Interestingly, the most common biologically relevant affected genes were retinoic acid receptors, thyroid hormone receptors, or hormone/nuclear receptors, important genes related to intracellular pathways and regulators of gene expression (9).
In summary, this large body of evidence supports the concept that prolonged GC exposure modulates the epigenetic landscape across the genome by inducing DMR and histone modifications. Some epigenetic modifications are persistent, and this may partially explain the incomplete reversibility of some of CS features following clinical remission.
Glucocorticoid-Target Genes in Cushing Syndrome
A detailed identification and characterization of GC-target genes may shed light in the understanding of the pathophysiology and treatment response in patients with CS. For instance, the GC regulation of pro-opiomelanocortin (POMC) expression as part of the physiologic GC negative feedback may be impaired in Cushing disease (CD), which is an important mechanism for the maintenance of high GC levels (53). Another example is the interaction between GC and clock genes, which may interfere in the loss of the GC circadian rhythm and may contribute to metabolic disorders in CS (54). Furthermore, the suppressive action of GC on drug targets, such as the somatostatin receptor (subtype 2), may influence the efficacy of first-generation somatostatin receptor ligands in normalizing cortisol levels in CD (55). Here we describe how GCs using epigenetic machinery influence the expression of important target genes and their implications in CS.
FKBP5
FK506 binding protein (FKBP5) plays an important role in the regulation of hypothalamic-pituitary-adrenal (HPA) system (56). As part of the GC negative feedback loop, GC binds to hypothalamic and pituitary GR. In the cytoplasm, GR is bound to a multi-protein complex including FKBP5. FKBP5 modulates GR action by decreasing GR binding affinity to GC and by preventing GR translocation from cytoplasm to nucleus (57, 58). In other words, an increase of FKBP5 expression is inversely correlated with GR activity and results in GC resistance leading to an impaired negative feedback regulation in the HPA axis (59).
FKBP5 is a GC-responsive gene; its upregulation by GC is part of an intracellular negative short-feedback loop (60). The mechanism by which GC regulates FKBP5 expression was shown to include inhibition of DNA methylation (44). In a model for CS, mice treated with corticosterone for 4 weeks had a reduced level of DNA methylation of FKBP5 in DNA extracted from whole blood, which was strongly correlated in a negative manner with GC concentration. Interestingly, a negative correlation was also observed between the degree of FKBP5 gene methylation measured at 4 weeks of GC exposure and the percentage of mice visceral fat (61). Accordingly, previous studies have provided compelling evidence of decreased methylation in the FKBP5 gene in patients with active CS compared to healthy control (10, 46). Even in patients with CS in remission, previous data have suggested a small decrease in FKBP5 methylation levels compared to healthy controls (9, 10). In an in vitro study, it was demonstrated that, by decreasing DNMT1 expression, GC is able to reduce FKBP5 methylation levels and, therefore, increase its expression (44).
Likewise, FKBP5 mRNA is also sensitive to GC exposure. A time-dependent increase in blood FKBP5 mRNA after single-dose prednisone administration has been demonstrated in healthy humans (62). Accordingly, patients with ACTH-dependent CS had higher blood FKBP5 mRNA levels compared with healthy controls, and after a successful surgery, FKBP5 mRNA returned to baseline levels (63). Furthermore, in another study, blood FKBP5 mRNA was inversely correlated with FKBP5 promoter methylation and positively correlated with 24-hour urine free cortisol (UFC) levels in patients with CS (46). Taken together, this fine-tuning of FKBP5 DNA methylation and mRNA according to the level of GC suggests that FKBP5 can be used as a biomarker to infer the magnitude of GC exposure.
POMC and Corticotropin-Releasing Hormone
The partial resistance of the corticotroph adenoma to GC negative feedback is a hallmark of CD. Indeed, the lack of this inhibitory effect constitutes a method to diagnose CD, that is, with the dexamethasone suppression test. One of the mechanisms related to the insensitivity to GC can be attributed to GR mutations which are, however, rarely found in corticotrophinomas (64). Another mechanism that was uncovered in corticotroph adenomas is an overexpression of the HSP90 chaperone resulting in reduced affinity of GR to its ligand and consequently GR resistance (53, 65).
In addition, the loss of protein expression of either Brg1, ATPase component of the SWI/SNF chromatin remodeling complex, or HDAC2 has been linked to GC resistance in about 50% of some adenomas (66). The trans-repression process on POMC transcription achieved by GC involves both the histone deacetylation enzyme and Brg1. One mechanism of corticotropin-releasing hormone (CRH)-induced POMC expression is through an orphan nuclear receptor (NR) related to NGFI-B (Nur77). NGFI-B binds to the NurRE sequence in the promoter region of POMC gene and recruits a co-activator to mediate its transcription. In a tethering mechanism, the GR directly interacts with NGFI-B to form a trans-repression complex, which contains the GR itself, Brg1, the nuclear receptor, and HDAC2; the latter being essential to block the gene expression through chromatin remodeling process (53, 66).
In CD, hypercortisolism exerts a negative feedback at CRH secretion from the hypothalamus (67). The mechanism involved in GR-induced suppression of CRH expression is through direct binding to a nGRE in the promoter region of CRH gene and subsequent recruitment of repressor complexes. In a rat hypothalamic cell line, it was demonstrated that Dex-induced CRH repression occurs through coordinated actions of corepressors involving Methyl-CpG-binding protein 2 (MeCP2), HDAC1, and DNA methyltransferase 3B (DNMT3B). Possibly, GR bound to nGRE recruits DNMT3B to the promoter in order to methylate a specific region, subsequently binding MeCP2 on these methylated sites followed by the recruitment of chromatin modify corepressor HDAC1, ultimately resulting in CRH suppression. Another possibility is that 2 independent complexes, one consisting of GR with DNMT3 for the methylation and the other the MeCP2, bound to methylated region, interact with HDAC1 to induce repression (68).
Clock Genes
The clock system and the HPA axis are interconnected regulatory systems. Cortisol circadian rhythm is modulated by the interaction between a central pacemaker, located in the hypothalamic suprachiasmatic nuclei, and the HPA axis (69). At the molecular level, mediators of the clock system and cortisol also communicate with each other, both acting as transcription factors of many genes to influence cellular functions.
In CS, the impact of chronic GC exposure on clock genes expression was recently evaluated using peripheral blood samples from patients with active disease compared with healthy subjects. The circadian rhythm of peripheral clock gene expression (CLOCK, BMAL, PER1-3, and CRY1) was abolished as a result of hypercortisolism, and that may contribute to metabolic disorders observed in Cushing patients (70). Another study, which investigated persistent changes induced by hypercortisolism in visceral adipose tissue, found that the expression of clock genes, such as PER1, remained altered in association with persistent epigenetic changes in both H3K4me3 and H3K27ac induced by hypercortisolism even after the resolution of hypercortisolism (12). This suggests that chronic exposure to GC may induce sustained epigenetic changes that can influence clock genes expression. Nevertheless, further studies are warranted to better elucidate how long-term exposure to GC impacts clock genes expression using the epigenetic machinery.
Glucocorticoid Effects on MicroRNAs
Along with histone modification and DNA methylation, microRNAs (miRNAs) have emerged as an epigenetic mechanism capable of impacting gene expression without changing DNA sequence (15). Interestingly, miRNA expression itself is also under the influence of epigenetic modifications through promoter methylation like any other protein-encoding genes (71).
MicroRNAs are small (about 20-25 nucleotides in length) non-coding RNAs that are important in transcriptional silencing of messenger RNA (mRNA). By partially pairing with mRNA, miRNAs can either induce mRNA degradation or inhibit mRNA translation to protein. MiRNAs regulate the translation of about 50% of the transcriptome, allowing them to play an important role in a wide range of biological functions, such as cell differentiation, proliferation, metabolism, and apoptosis under normal physiological and pathological situations. Some miRNAs can be classified as oncogenes or tumor suppressing genes, and aberrant expression of miRNAs may be implicated in tumor pathogenesis (71-73).
Insight into the regulation of miRNA expression is, therefore, crucial for a better understanding of tumor development and other human diseases, including cardiac, metabolic, and neurological disorders (73, 74). There are different regulatory mechanisms involved in miRNA expression, including transcriptional factors such as GR-GC. GC may modulate miRNA expression through direct binding to GRE in the promoter region of the host gene, as observed in hemopoietic tumor cells (75). In addition to transcriptional activation, in vascular smooth muscle cells, Dex treatment induces downregulation of DNMT1 and DNMT3a protein levels and reduces the methylation of miRNA-29c promoter, resulting in an increased expression of miRNA-29c (76). Interestingly, it was demonstrated that the increased expression of miRNA-29 family (miRNA-29a, -29b, and -29c) associates with metabolic dysfunction, such as obesity and insulin resistance, which pertains to CS (77, 78). With regard to metabolic dysfunction, miRNA-379 expression was shown to be upregulated by GC and its overexpression in the liver resulted in elevated levels of serum triglycerides associated with very low-density lipoprotein (VLDL) fraction in mice (79). In obese patients, the level of hepatic miRNA-379 expression was higher compared to nonobese patients and positively correlated with serum cortisol and triglycerides (79). Hence, GC-responsive miRNA may be, at least in part, a mediator to GC-driven metabolic conditions in CS.
In pathological conditions, such as seen in CS, prolonged exposure to an elevated cortisol level results in a wide range of comorbidities. It can be hypothesized that the chronic and excessive glucocorticoid levels may induce an aberrant miRNA expression that might impact several cellular processes related to bone and cardiometabolic disorders. A recent study addressed the impact of hypercortisolism on bone miRNA of patients with active CD compared to patients with nonfunctional pituitary adenomas. Significant changes in bone miRNA expression levels were observed, suggesting that the disruption of miRNA may be partially responsible for reduced bone formation and osteoblastogenesis (80). Similarly, altered expression levels of selected miRNAs related to endothelial biology in patients with CS may point to a contribution to a high incidence of cardiovascular disorders in Cushing patients (81). Therefore, dysregulated miRNAs as a consequence of high cortisol levels may underpin the development and progression of comorbidities related to CS. To the best of our knowledge, it is currently not clear whether miRNA dysregulation persists after resolution of hypercortisolism, thus contributing to the persistence of some comorbidities. This hypothesis needs to be further investigated.
MicroRNA can also be used as a diagnostic tool in CS. A study was performed to identify circulating miRNA as a biomarker to differentiate patients with CS from patients with suspected CS who had failed diagnostic tests (the control group) (82). It was observed that miRNA182-5p was differentially expressed in the CS cohort compared to the control group; therefore, it may be used as a biomarker (82). However, a large cohort is necessary to validate this finding (82). In corticotroph tumors, downregulation of miRNA 16-1 expression was observed relative to normal pituitary tissue (83). In contrast, the plasma level of miRNA16-5p was found to be significantly higher in CD compared to ectopic Cushing (EAS) and healthy controls (84). This finding suggests that miRNA16-5p may be a biomarker capable to differentiate the 2 forms of ACTH-dependent Cushing (84).
Epidrugs and Glucocorticoid Action in Cushing's Syndrome
The interest in understanding the epigenetic mechanism of GC action in the context of CS is based on reversibility of epi-marks, such as DNA methylation and histone modifications, using epidrugs (85, 86). The biological characteristics of epigenetic drugs and their target have been extensively explored. Their effectiveness as antitumor drugs have been tested on corticotroph tumors using in vitro studies (87-89). However, a limited number of studies have explored the role of epidrugs as a therapeutic tool in reversing the genomic action of GC in CS, particularly in comorbidities induced by hypercortisolism (90, 91).
The use of histone deacetylase inhibitors (HDACi) may reduce the genomic action of GC (90-92). It has been demonstrated that the use of the HDAC inhibitor valproic acid increases the acetylation level of GR, consequently attenuating the genomic action of GC. In an experimental Cushing model in rats, the use of valproic acid decreased expression of genes related to lipogenesis, gluconeogenesis, and ion regulators in the kidney that ultimately reduces hepatic steatosis, hyperglycemia, and hypertension in ACTH-infused rats (90, 91).
More studies evaluating the effects of epidrugs influencing the GC actions are warranted to further elucidate the underlying mechanisms and to explore potential treatment modalities to reverse long-lasting consequences of chronic corticoid exposure.
Conclusions
In physiologic conditions, GC are secreted in pulses following a circadian rhythm pattern, as opposed to a constant, chronic, and high GC exposure in CS. This pathological pattern may account for numerous devastating effects observed in CS (7). Yet, the expressed genome in response to chronic GC exposure may potentially be abnormal, leading to dysregulation in clock genes, among other effects.
GC levels may return to a normal circadian pattern in response to a successful treatment, but with incomplete reversibility of some CS features, which may in part be explained by epigenetic changes. The epigenetic machinery is used by GC to induce dynamic changes in chromatin to modulate gene expression. (Fig. 2) It seems that most of chromatin modifications are reversible, but some may persist resulting in long-term epigenetic changes. (Table 1)
Table 1.Evidence of interaction between glucocorticoid and epigenetic machinery
Epigenetic changes/epigenetic enzymes Action Histone acetylation (HAT) Histone deacetylation (HDAC) -
GR recruit histone deacetylases (HDACs) to turn chromatin less accessible and suppress gene transcription (25, 35).
-
The trans-repression process on POMC transcription achieved by glucocorticoids (GC) involves the histone deacetylation enzyme (HDAC2).
-
GC mediates the upregulation of HDAC2 in rats exposed to chronic stress (40).
Histone demethylase (JMJD3) -
GC suppress transcription of JMJD3 in endothelial cells treated with TNFα (41).
Histone modifications -
Using ChIP-seq, a study in mice treated for 5 weeks with corticosterone showed higher levels of histone modifications (H3K4me3, H3K27ac) compared to control mice. In mice after a 10-week washout period, persistence of this epigenetic fingerprint was observed, which was associated with long-lasting changes in gene expression (12).
DNA methylation (DNMT3B) and histone deacetylation (HDAC1) -
GC mediates CRH downregulation through DNMT3B to the promoter in order to methylate a specific region and recruitment of chromatin modify corepressor HDAC (68).
DNA hypomethylation -
GC induces downregulation of DNMT1 in AtT20 (mouse corticotroph adenoma cell line) (20).
-
GC induces upregulation of TET enzyme expression which was described in retinal and osteocyte cell line model (42, 43).
-
An experimental study in mice previously exposed to high levels of GC showed differentially methylated regions (DMR) induced by GC treatment, of which the majority was loss of the methylation (11).
-
Reduced DNA methylation in FKBP5 gene was found in patients in active disease and also in remission state of Cushing syndrome (CS) as compared to a healthy control group (10).
-
A genome-wide DNA methylation analysis showed a lower average of DNA methylation in patients in remission of CS compared to controls (9).
-
A study using whole blood methylation profile demonstrated an association between cortisol excess and DNA hypomethylation in patients with CS (46).
Further studies are needed to elucidate how chronic exposure to GC leads to incomplete reversibility of CS morbidities via sustained modulation of the epigenetic machinery and possibly other mechanisms. Subsequent identification of therapeutic targets may offer new perspective for treatments, for example, with epidrugs, aiming to reverse hypercortisolism-related comorbidities.
Funding
The authors received no financial support for this manuscript.
Disclosures
T.P., R.A.F., and L.J.H. have nothing to declare.
Data Availability
Data sharing is not applicable to this article, as no datasets were generated or analyzed during the current study.
-
 1
1
-
-
Wow - it's nearly 18 years since my cancer surgery.
So, I've been back on the growth hormone for about 8 years. I still don't feel like it's doing much/any good but it must be since tests come out ok, my endo still prescribes it and my insurance still pays for a huge chunk of it.
~~~
Cushing's awareness discussions come out in the oddest ways.
Last night, we were out at a Lebanese restaurant couple with a young couple from India and were talking about foods we liked. I said that I liked spicy foods in general.
That led to the fact that I haven't had a good sense of smell since my transsphenoidal surgery and that things taste better when I can smell them. That got us around to why I'd had the surgery and what the symptoms of Cushing's are.
Our dinner companions will probably never hear of Cushing's again but if they do, they'll know what it is!

-
And then there were two
")

-
Abstract
Cushing’s syndrome is a constellation of features occurring due to high blood cortisol levels. We report a case of a 47-year-old male with a history of recurrent olfactory neuroblastoma (ONB). He presented with bilateral lower limb weakness and anosmia and was found to have Cushing’s syndrome due to high adrenocorticotropic hormone (ACTH) levels from an ectopic source, ONB in this case. Serum cortisol and ACTH levels declined after tumor removal.
Introduction
Olfactory neuroblastoma (ONB), or esthesioneuroblastoma, is a rare malignancy arising from neuroepithelium in the upper nasal cavity. It represents approximately 2% of all nasal passage tumors, with an incidence of approximately 0.4 per 2.5 million individuals [1]. ONB shares similar histological features with small round blue cell neoplasms of the nose. Ectopic hormone secretion is a very rare feature associated with these tumors. Five-year overall survival is reported to be between 60% and 80% [2,3]. The age distribution is either in the fifth to sixth decade of life [4,5], or in the second and sixth decades [6].
Features of Cushing’s syndrome (moon face, buffalo hump, central obesity hypertension, fragile skin, easy bruising, fatigue, muscle weakness) are due to high blood cortisol levels [7]. It can be either primary (cortisol-secreting adrenal tumor), secondary (adrenocorticotropic hormone (ACTH)-secreting pituitary tumor, also called Cushing disease), or ectopic ACTH secretion (from a non-pituitary source). All three types share similar features [8].
Ectopic ACTH syndrome (EAS) is due to an extra pituitary tumor, producing ACTH. It accounts for 12-17% of Cushing's syndrome cases [9]. Most cases of EAS-producing tumors are in the lungs, mediastinum, neuroendocrine tumors of the gastrointestinal tract, and pheochromocytomas [9]. Ectopic ACTH secretion from an ONB is very rare. As of 2015, only 18 cases were reported in the literature [10]. Here, we report such a case.
Case Presentation
Our patient is a 47-year-old Bangladeshi male, with a history of recurrent ONB that was resected twice in the past (transsphenoidal resection in 2016 and 2019) with adjuvant radiotherapy, no chemotherapy was given. He also had diabetes mellitus type 1 (poorly controlled) and hypertension. He presented with bilateral lower limb weakness, anosmia, decreased oral intake, loss of taste for one week, and bilateral submandibular swelling that increased in size gradually over the past two years. There was no history of fever, cough, abdominal pain, or exposure to sick contacts. The patient reported past episodes of similar symptoms, but details are unclear. The patient's family history is positive for diabetes mellitus type 1 in both parents. Lab tests in the emergency department showed hypokalemia and hyperglycemia as detailed in Table 1. He was admitted for further workup of the above complaints.
Test Patient Results Reference Range Unit Status Hemoglobin 14.7 13-17 g/dL Normal White blood cell (WBC) 17.9 4-10 10*9/L High Neutrophils 15.89 2-7 10*9/L High Lymphocytes 1.07 1-3 10*9/L Normal Sodium 141 136-145 mmol/L Normal Potassium 2.49 3.5-5.1 mmol/L Low (Panic) Chloride 95 98-107 mmol/L Low Glucose 6.52 4.11-5.89 mmol/L Elevated C-reactive protein (CRP) 0.64 Less than 5 mg/L Normal Erythrocyte sedimentation rate (ESR) 2 0-30 mm/h Normal Creatinine 73 62-106 µmol/L Normal Uric acid 197 202.3-416.5 µmol/L Normal Alanine aminotransferase (ALT) 33.2 0-41 U/L Normal Aspartate aminotransferase (AST) 18.6 0-40 U/L Normal International Normalised Ratio (INR) 1.21 0.8-1.2 sec High Prothrombin time (PT) 15.7 12.3-14.7 sec High Lactate dehydrogenase (LDH) 491 135-225 U/L High Thyroid-stimulating hormone (TSH) 0.222 0.27-4.20 mIU/L Low Adrenocorticotropic hormone (ACTH) 106 ≤50 ng/L Elevated Cortisol (after dexamethasone suppression) 1750 Morning hours (6-10 am): 172-497 nmol, Afternoon hours (4-8 pm): 74.1-286 nmol nmol/L Elevated (failure of suppression) 24-hour urine cortisol (after dexamethasone suppression) 5959.1 <120 nmol/24 hrs nmol/24hr Elevated (failure of suppression) Table 1: Results of blood test at the time of hospitalization. Hypokalemia and high values of adrenocorticotropic hormone and cortisol were confirmed.
On examination, the patient's vital signs were as follows: blood pressure was 154/77 mmHg, heart rate of 60 beats per minute, respiratory rate was 18 breaths per minute, oxygen saturation of 98% on room air, and a temperature of 36.7°C. The patient had a typical Cushingoid appearance with a moon face, buffalo hump, purple striae on the abdomen, central obesity, and hyperpigmentation of the skin. Submandibular lymph nodes were enlarged bilaterally. The examination of the submandibular lymph nodes showed a firm, fixed mass extending from the angle of the mandible to the submental space on the left side. Neurological examination showed weakness in both legs bilaterally (strength 3/5) and anosmia (checked by orthonasal smell test). The rest of the neurological exam was normal.
Laboratory findings revealed (in Table 1) a marked hypokalemia of 2.49 mmol/L and hyperglycemia of 6.52 mmol/L. The serum cortisol level was elevated at 1587 nmol/L. Serum ACTH levels were raised at 106 ng/L (normal value ≤50 ng/L). Moreover, the high-dose dexamethasone suppression test failed to lower the serum ACTH levels and serum and urine cortisol. Serum cortisol level after the suppression test was 1750 nmol/L, while 24-hour urine cortisol after the test was 5959.1 nmol/24hr. Serum ACTH levels after the test also remained high at 100mg/L. This indicated failure of ACTH suppression by high-dose dexamethasone, which points towards ectopic ACTH production. Other blood tests (complete blood count, liver function tests) were insignificant.
A computed tomography scan with contrast (CT scan) of the chest, abdomen, and pelvis, with a special focus on the adrenals, was negative for any malignancy or masses. CT scan of the neck showed bilaterally enlarged submandibular lymph nodes and an enlarged right lobe of the thyroid with nodules. Fine needle aspiration (FNA) of the thyroid nodules revealed a benign nature. Magnetic resonance imaging (MRI) of the brain showed a contrast-enhancing soft tissue lesion (18x18x10mm) in the midline olfactory groove area with extension into the frontal dura and superior sagittal sinus, suggesting recurrence of the previous ONB. There was evidence of previous surgery also. The pituitary gland was normal (Figures 1-2).
-shows-a-soft-tissue-lesion-located-in-the-midline-olfactory-groove-area.-Dural-surface-with-extension-into-anterior-frontal-dura.")
Figure 1: A brain MRI (T1-weighted; without contrast; sagittal plane) shows a soft tissue lesion located in the midline olfactory groove area. Dural surface with extension into anterior frontal dura.
MRI: Magnetic resonance imaging
-shows-a-soft-tissue-lesion-located-in-the-midline-olfactory-groove-area.")
Figure 2: A brain MRI (T2-weighted; without contrast; axial plane) shows a soft tissue lesion located in the midline olfactory groove area.
MRI: Magnetic resonance imaging
Octreotide scintigraphy showed three focal abnormal uptakes in the submandibular cervical nodes. Additionally, there was a moderate abnormal uptake at the midline olfactory groove with bilateral extension (Figure 3).
-demonstrates-three-focal-abnormal-uptakes:-the-largest-(5.2-x-2.4-cm)-in-the-left-submandibular-region,-and-two-smaller-ones-on-the-right,-suggestive-of-lymph-node-uptake.-Additional-abnormal-uptake-was-seen-along-the-midline-of-the-olfactory-groove-region-with-bilateral-extension.-No-other-significant-abnormal-uptake-was-identified.")
Figure 3: Whole-body octreotide scan (15 mCi 99mTc-Octreotide IV) demonstrates three focal abnormal uptakes: the largest (5.2 x 2.4 cm) in the left submandibular region, and two smaller ones on the right, suggestive of lymph node uptake. Additional abnormal uptake was seen along the midline of the olfactory groove region with bilateral extension. No other significant abnormal uptake was identified.
On microscopic examination, an excisional biopsy after the transcranial resection surgery of the frontal skull base tumor showed nests and lobules of round to oval cells with clear cytoplasm, separated by vascular and hyalinized fibrous stroma (Figures 4A-4B). Tumor cells show mild to moderate nuclear pleomorphism, and fine chromatin (Figure 4C). A fibrillary neural matrix is also present. Some mitotic figures can be seen. Immunohistochemical stains revealed positive staining for synaptophysin (Figure 4D) and chromogranin (Figure 4E). Stains for CK (AE1/AE3), CD45, Desmin, and Myogenin are negative. Immunostaining for ACTH was focally positive (Figure 4F), while the specimen of the cervical lymph nodes showed the same staining, indicating metastases. The cytomorphologic and immunophenotypic features observed are consistent with a Hyams grade II ONB, with ectopic ACTH production.

Figure 4: Histopathological and immunohistochemical findings of olfactory neuroblastoma.
A (100x magnification) and B (200x magnification) - hematoxylin and eosin (H-E) staining shows cellular nests of round blue cells separated by hyalinized stroma. C (400x magnification) - nuclei show mild to moderate pleomorphism with fine chromatin. D (100x magnification) - an immunohistochemical stain for synaptophysin shows diffuse, strong cytoplasmic positivity within tumor cells. E (200x magnification) - tumor cells are positive for chromogranin. F (400x magnification) - ACTH cytoplasmic expression in tumor cells.
ACTH: adrenocorticotropic hormone
For his resistant hypokalemia, he had to be given intravenous (IV) and oral potassium chloride (KCL) repeatedly. The patient underwent transcranial resection of the frontal skull base tumor. The patient received cefazolin for seven days, and hydrocortisone for four days. After transcranial resection, his cortisol level decreased to 700 nmol/L. Furthermore, ACTH dropped, and serum potassium also normalized. Subsequently, the patient was transferred to the intensive care unit (ICU) for meticulous monitoring and continued care. In the ICU, the patient developed one episode of a generalized tonic-clonic seizure, which aborted spontaneously, and the patient received phenytoin and levetiracetam to prevent other episodes. A right-sided internal jugular vein and left transverse sinus thrombosis were also developed and treated with enoxaparin sodium. Following surgery, his low potassium levels improved, resulting in an improvement in his limb weakness. His other symptoms also gradually improved after surgery. Three weeks following the primary tumor resection, he underwent bilateral neck dissection with right hemithyroidectomy, for removal of the metastases. The patient opted out of chemotherapy and planned for an international transfer to his home country for further management. Other treatments that he received during hospitalization were ceftriaxone, azithromycin, and Augmentin®. Insulin was used to manage his diabetes, perindopril to regulate his blood pressure, and spironolactone to increase potassium retention. Omeprazole was administered to prevent GI bleeding and heartburn/gastroesophageal reflux disease relief after discharge.
Discussion
ONB was first described in 1924, and it is a rare neuroectodermal tumor that accounts for 2% of tumors affecting the nasal cavity [11]. Even though ONB has a good survival rate, long-term follow-up is necessary due to the disease's high recurrence rate [2]. ONB recurrence has been approximated to range between 30% and 60% after successful treatment of the primary tumor [12]. Recurrent disease is usually locoregional and tends to have a long interval to relapse with a mean of six years [12]. The first reported case of ectopic ACTH syndrome caused by ONB was in 1987 by M Reznik et al., who reported a 48-year-old woman with ONB who developed a Cushing-like syndrome 28 months before her death [13].
The occurrence of Cushing’s syndrome due to ectopic ACTH can occur either in the initial tumor or even years later during its course or after recurrence [3,6,9,14]. Similar to the case of Abe et al. [3], our patient also presented with muscle weakness due to hypokalemia, which is a feature of Cushing’s syndrome. Hypokalemia is present at diagnosis in 64% to 86% of cases of EAS and is resistant to treatment [9,14], as seen in our case. In our patient, the exact time of development of Cushing’s syndrome could not be ascertained due to the non-availability of previous records. However, according to the patient, he started developing abdominal obesity, pigmentation, and buffalo hump in 2021 about two years after his second surgery for ONB.
The distinction between pituitary ACTH and ectopic ACTH involves utilizing CT/MRI of the pituitary, corticotropin-releasing hormone (CRH) stimulation test with petrosal sinus blood sampling, high dose dexamethasone suppression test, and checking serum K+ (more commonly low in ectopic ACTH) [2,15,16]. In our case, a CRH stimulation test was not available but CT/MRI brain, dexamethasone test, low serum potassium, plus the postoperative fall in cortisol levels, all pointed towards an ectopic ACTH source.
Conclusions
In conclusion, this case highlights the rare association between ONB and ectopic ACTH syndrome, which developed after tumor recurrence. The patient's unique presentation of bilateral lower limb weakness and hypokalemia can cause diagnostic challenges, emphasizing the need for comprehensive diagnostic measures. Surgical intervention proved crucial, with postoperative cortisol values becoming normal, highlighting the efficacy of this approach. The occurrence of ectopic ACTH production in ONB patients, although very rare, is emphasized, so that healthcare professionals who deal with these tumors are aware of this complication. This report contributes valuable insights shedding light on the unique ONB manifestation causing ectopic ACTH syndrome. The ongoing monitoring of the patient's clinical features will further enrich the understanding of the course of this uncommon phenomenon in the medical literature.
References
- Thompson LD: Olfactory neuroblastoma. Head Neck Pathol. 2009, 3:252-9. 10.1007/s12105-009-0125-2
- Abdelmeguid AS: Olfactory neuroblastoma. Curr Oncol Rep. 2018, 20:7. 10.1007/s11912-018-0661-6
- Abe H, Suwanai H, Kambara N, et al.: A rare case of ectopic adrenocorticotropic hormone syndrome with recurrent olfactory neuroblastoma. Intern Med. 2021, 60:105-9. 10.2169/internalmedicine.2897-19
- Yin Z, Wang Y, Wu Y, et al.: Age distribution and age-related outcomes of olfactory neuroblastoma: a population-based analysis. Cancer Manag Res. 2018, 10:1359-64. 10.2147/CMAR.S151945
- Platek ME, Merzianu M, Mashtare TL, Popat SR, Rigual NR, Warren GW, Singh AK: Improved survival following surgery and radiation therapy for olfactory neuroblastoma: analysis of the SEER database. Radiat Oncol. 2011, 6:41. 10.1186/1748-717X-6-41
- Elkon D, Hightower SI, Lim ML, Cantrell RW, Constable WC: Esthesioneuroblastoma. Cancer. 1979, 44:3-1087. 10.1002/1097-0142(197909)44:3<1087::aid-cncr2820440343>3.0.co;2-a
- Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, Montori VM: The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008, 93:1526-40. 10.1210/jc.2008-0125
- Chabre O: Cushing syndrome: physiopathology, etiology and principles of therapy [Article in French]. Presse Med. 2014, 43:376-92. 10.1016/j.lpm.2014.02.001
- Isidori AM, Lenzi A: Ectopic ACTH syndrome. Arq Bras Endocrinol Metabol. 2007, 51:1217-25. 10.1590/s0004-27302007000800007
- Kunc M, Gabrych A, Czapiewski P, Sworczak K: Paraneoplastic syndromes in olfactory neuroblastoma. Contemp Oncol (Pozn). 2015, 19:6-16. 10.5114/wo.2015.46283
- Finlay JB, Abi Hachem R, Jang DW, Osazuwa-Peters N, Goldstein BJ: Deconstructing olfactory epithelium developmental pathways in olfactory neuroblastoma. Cancer Res Commun. 2023, 3:980-90. 10.1158/2767-9764.CRC-23-0013
- Ni G, Pinheiro-Neto CD, Iyoha E, et al.: Recurrent esthesioneuroblastoma: long-term outcomes of salvage therapy. Cancers (Basel). 2023, 15:1506. 10.3390/cancers15051506
- Reznik M, Melon J, Lambricht M, Kaschten B, Beckers A: Neuroendocrine tumor of the nasal cavity (esthesioneuroblastoma). Apropos of a case with paraneoplastic Cushing's syndrome [Article in French]. Ann Pathol. 1987, 7:137-42.
- Kadoya M, Kurajoh M, Miyoshi A, et al.: Ectopic adrenocorticotropic hormone syndrome associated with olfactory neuroblastoma: acquirement of adrenocorticotropic hormone expression during disease course as shown by serial immunohistochemistry examinations. J Int Med Res. 2018, 46:4760-8. 10.1177/0300060517754026
- Clotman K, Twickler MTB, Dirinck E, et al.: An endocrine picture in disguise: a progressive olfactory neuroblastoma complicated with ectopic Cushing syndrome. AACE Clin Case Rep. 2017, 3:278-83. 10.4158/EP161729.CR
- Chung YS, Na M, Ku CR, Kim SH, Kim EH: Adrenocorticotropic hormone-secreting esthesioneuroblastoma with ectopic Cushing’s syndrome. Yonsei Med J. 2020, 61:257-61. 10.3349/ymj.2020.61.3.257
-
1
-
You know, I'm still not sure if the GH is worth it, even after almost 20 years (with some hiccups). I had to stop after I've had kidney cancer and flying with the stuff is such a pain due to the refrigeration (more about that here).
One time we were going on a cruise out of New York so we were visiting our son first. I was in a hotel and I put the whole case in an ice bucket. The ice melted... I wasn't sure if the water had seeped into the injector pen or not so I threw it away and went on the cruise without it. I never noticed any bad effects from the week or so with no GH.
So, last January when we went on another cruise, I didn't bother to take it with no ill effects.
So, it doesn't seem like it's working for me but my endo (Dr. Salvatori at Johns Hopkins) is happy so I guess my non-schedule is ok. Maybe I'll just die sooner than I would have.

-
1
-
-
Authors Stasiak M , Witek P, Adamska-Fita E, Lewiński A
Received 27 December 2023
Accepted for publication 20 March 2024
Published 8 April 2024 Volume 2024:16 Pages 35—42
DOI https://doi.org/10.2147/DHPS.S453105
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Hemalkumar B Mehta
Magdalena Stasiak,1 Przemysław Witek,2 Emilia Adamska-Fita,1 Andrzej Lewiński1,3
1Department of Endocrinology and Metabolic Diseases, Polish Mother’s Memorial Hospital—Research Institute, Lodz, Poland; 2Department of Internal Medicine, Endocrinology and Diabetes, Medical University of Warsaw; Mazovian Brodnowski Hospital, Warszawa, Poland; 3Department of Endocrinology and Metabolic Diseases, Medical University of Lodz, Lodz, Poland
Correspondence: Magdalena Stasiak, Department of Endocrinology and Metabolic Diseases, Polish Mother’s Memorial Hospital—Research Institute, 281/289 Rzgowska Street, Lodz, 93-338, Poland, Tel +48502049292, Fax +48422711140, Email mstasiak33@gmail.com
Abstract: Cushing’s disease (CD) is the most common cause of endogenous hypercortisolism. Osilodrostat was demonstrated to be efficient in treating CD, and the mean average dose required for CD control was < 11 mg/day. Potential differences in osilodrostat treatment between cortisol-producing adenoma (CPA) and CD have not been reported. The aim of this study was to present two patients with CPA in whom significant differences in the response to therapy compared to CD were found. We demonstrated a case of inverse response of cortisol levels with adrenal tumor progression during the initial dose escalation (Case 1). Simultaneously, severe exaggeration of hypercortisolism symptoms and life-threatening hypokalemia occurred. A further rapid dose increase resulted in the first noticeable cortisol response at a dose of 20 mg/day, and a full response at a dose of 45 mg/day. We also present a case that was initially resistant to therapy (Case 2). The doses required to achieve the first response and the full response were the same as those for Case 1. Our study demonstrated that osilodrostat therapy in patients with CPA may require a different approach than that in CD, with higher doses, faster dose escalation, and a possible initial inverse response or lack of response.
Keywords: osilodrostat, adrenal adenoma, hypercortisolism, ACTH-independent, adverse events, hypokalemiaIntroduction
Chronic persistent hypercortisolism is a life-threatening condition that requires effective treatment. Untreated exposure to excessive cortisol secretion leads to severely increased morbidity and mortality due to cardiovascular diseases, thromboembolic events, sepsis, visceral obesity, impairment of glucose metabolism, and dyslipidaea, as well as musculoskeletal disorders, such as myopathy, osteoporosis, and skeletal fractures. Moreover, neuropsychiatric disorders, such as impairment of cognitive function, depression, or mania, as well as impairment of reproductive function can frequently occur.1,2 Cushing’s disease (CD) – a disorder caused by a pituitary adenoma secreting adrenocorticotropic hormone (ACTH) – is the most common cause of hypercortisolism. Cushing’s syndrome (CS) includes all other causes of cortisol excess, including ectopic ACTH production as well as direct cortisol overproduction by adrenal adenoma (cortisol-producing adenoma [CPA]) or adrenocortical carcinoma (ACC). Approximately 10% of hypercortisolism cases result from CPA. The first line therapy is a surgical resection of the tumor, which is the source of hormone excess. However, in many patients surgery is not fully efficient and other therapies are required to reduce cortisol levels. Additionally, due to severe cardiovascular complications and unstable DM, the surgical approach sometimes entails unacceptable risk and it is frequently postponed until cortisol levels are lowered. Pharmacotherapy with steroidogenesis inhibitors reduces cortisol levels and improves the symptoms of hypercortisolism.1,2 As CD is the most common cause of cortisol excess, most studies have focused on the efficacy and safety of novel steroidogenesis inhibitors, including patients with CD only.3–6 This is exactly the case with osilodrostat – a new potent inhibitor of 11β-hydroxylase.3–6 More data are available for metyrapone efficacy and safety in CSA,7 as the drug has been available much longer than osilodrostat. A study by Detomas et al, which reported results of comparison of efficacy of metyrapone and osilodrostat, included 4 patients with adrenal CS, among whom one CPA patient was treated with osilodrostat.8 Osilodrostat is approved in the United States to treat CD in patients in whom pituitary surgery was not curative or is contraindicated.9 In Poland, osilodrostat therapy is available for patients with all kinds of endogenous hypercortisolism not curative with other approaches, within a national program of emergency access to drug technologies.10 Reports on osilodrostat application in CPA are highly valuable as data on potential differences in the treatment regimens between CD and CPA are scarce.
Here, we present two patients with CPA in whom the response and doses of osilodrostat were different from those reported in patients with CD. The main purpose of this study was to demonstrate that the efficacy of osilodrostat in CPA is high, although initial resistance to treatment or even deterioration of hypercortisolism can occur during the application of lower doses of the drug.
Materials and Methods
Study Design and Patients
We retrospectively analyzed medical files of two consecutive patients with CPA treated with osilodrostat. The analysis included medical history, laboratory and imaging results as well as a detailed reports of adverse events.
Laboratory and Imaging Procedures
Serum cortisol and ACTH levels were measured by electrochemiluminescence immunoassay (ECLIA) using a Cobas e601 analyzer (Roche Diagnostics, Indianapolis, IN, USA). UFC excretion was measured by chemiluminescent microparticle immunoassay (CMIA) using an Abbott Architect ci4100 analyzer (Abbott, Abbott Park, IL, USA). Cross-reactivity with 11-deoxycortisol for this method is very low (2.1% according to the manufacturer’s data). Potassium levels were measured by ion-selective electrode potentiometry using a Beckman Coulter DxC 700 AU Chemistry Analyzer (Beckman Coulter, Brea, CA, USA). Computed tomography (CT) imaging was performed using a Philips Ingenuity Core 128 system (Philips, the Netherlands).
Ethics Procedures
Informed consent was obtained from all subjects involved in the study. Written informed consent was obtained from the patients for publication of this paper. The approval of Institutional Ethics Committee was obtained to publish the case details (approval code KB 33/2023).
Presentation of the Cases
Case 1
A 51-year-old female was referred to our department in November 2021 because of CPA, disqualified from surgery because of severe hypertension with a poor response to antihypertensive therapy and uncontrolled DM despite high doses of insulin. Additionally, the patient presented with hyperlipidemia and severe obesity (BMI=50.7 kg/m2), gastritis, depression, and osteoarthritis. On admission, she complained of a tendency to gain weight, fragile skin that bruised easily, difficulty with wound healing, susceptibility to infections, and insomnia. Physical examination revealed a moon face with plethora, a buffalo hump, central obesity with proximal muscle atrophy, and purple abdominal striae.
The CPA diagnosis was initially made two years earlier, but the patient did not qualify for surgery due to a hypertensive crisis. Soon after this episode, the SARS-CoV-2 pandemic began, and the patient was afraid of visiting any medical center because her son had died of COVID-19. Therefore, she was referred to our center for life-threatening hypercortisolism two years later.
At the time of admission, computed tomography (CT) imaging revealed a right adrenal tumor of 34x24x37mm, with a basal density of 21 HU and a contrast washout rate typical for adenomas (83%). The size and CT characteristics were identical as they were two years earlier. High serum cortisol levels, undetectable ACTH concentrations, and a lack of physiological diurnal rhythm of cortisol secretion were observed (Table 1). Urinary free cortisol (UFC) excretion was 310 µg/24 h, with an upper normal limit (UNL) of 176 µg/24 h. No cortisol suppression was achieved in high-dose dexamethasone suppression test (DST) (Table 1). Other adrenal-related hormonal parameters were within normal ranges, with values as follows: DHEA-S 42.68 µg/dl, aldosterone 3.24 ng/mL, and renin 59.14 µIU/mL.

Table 1 Laboratory Results Before Osilodrostat Therapy – Case 1
Due to multiple severe systemic complications, including uncontrolled hypertension, decompensated DM, and cardiac insufficiency, treatment with osilodrostat was introduced for life-saving pre-surgical management. Osilodrostat was started at a dose of 1 mg twice daily and gradually increased to 6 mg per day with actually an inverse response of serum cortisol level. The late-night cortisol level increased from 16 µg/dl to 25 µg/dl. As the full effect of the osilodrostat dose can occur even after a few weeks, the patient was discharged from hospital and instructed to contact her attending doctor immediately if any health deterioration was noticed. In the case of improvement in the patient’s condition, the next hospitalization was planned 3 weeks later. After three weeks of no contact with the patient, she was readmitted to our department with life-threatening escalation of hypercortisolism, severe hypokalemia, and further deterioration of hypertension, DM, cardiac insufficiency, dyspnea, and significant edemas, including facial edema. Treatments of hypertension, cardiac insufficiency, and DM were intensified, as presented in Table 2. Despite active potassium supplementation, life-threatening hypokalemia of 2.1 mmol/l occurred. Previously observed depression was exaggerated with severe anxiety and fear of death. The dose of osilodrostat was increased to 8 mg/day, and after three days of treatment a further elevation of serum cortisol was found, with an increase in UFC up to 9 × UNL (1546.2 µg/24 h). Due to an entirely unexpected inverse cortisol response, CT imaging was performed and revealed progression of the adenoma size to 39 × 36 × 40 mm, with a slight increase in density up to 27 HU as compared to the previous CT scan performed a month earlier (Figure 1).

Table 2 Changes in the Most Important Parameters During Osilodrostat Therapy – Case 1
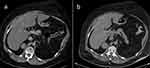
Figure 1 Progression of the adrenal adenoma size during the initial doses of osilodrostat: (a) CT scan directly before osilodrostat therapy – solid nodule 34x24x37 mm, basal density 21 HU; (b) CT scan during treatment with 8 mg of osilodrostat daily – solid nodule 39x36x40 mm, basal density of 27 HU.
Considering the extremely high risk associated with such a rapid cortisol increase and related complications, decision of fast osilodrostat dose escalation was made. The dose was increased by 5 mg every other day, up to 45 mg per day, and, finally, a gradual decrease in the cortisol level (Table 2) was achieved, with UFC normalization to 168 µg/24 h. During dose escalation, no deterioration in the adverse effects (AEs) of osilodrostat was observed. Conversely, hypokalemia gradually improved despite a simultaneous reduction in potassium supplementation (Table 2). Facial edema decreased and the level of anxiety improved significantly. The course of hypertension severity as well as a summary of the main parameters controlled during treatment and the medications used are presented in Table 2. As soon as the cortisol level normalized, the patient was referred for surgery and underwent right adrenalectomy without any complications. Histopathology results confirmed a benign adenoma of the right adrenal gland (encapsulated, well-circumscribed tumor consisting of lipid-rich cells with small and uniform nuclei, mostly with eosinophilic intracytoplasmic inclusions). After surgery, hydrocortisone replacement therapy was administered. A few days after surgery, blood pressure and glucose levels gradually decreased, and the patient required reduction of antihypertensive and antidiabetic medications. After 22 months of follow-up, the patient’s general condition is good with no signs of recurrence. Antidepressant treatment is no longer required in this patient. Body mass index was significantly reduced to 40 kg/m2. The antihypertensive medication was completely discontinued, and the glucose level is controlled only with metformin. The patient still requires hydrocortisone substitution at a dose of 30 mg/day.
Case 2
A 39-year-old female was referred to our department in November 2022 with a diagnosis of CPA and unstable hypertension, for which surgery was contraindicated. The patient was unsuccessfully treated with triple antihypertensive therapy (telmisartan 40 mg/day, nebivolol 5 mg/day, and lercanidipine 20 mg/day). The patient reported weight gain, muscle weakness, acne, fragile skin that bruised easily, and secondary amenorrhea. Other comorbidities included gastritis, hypercholesterolemia, and osteoporosis. Physical examination revealed typical signs of Cushing’s syndrome, such as abnormal fat distribution, particularly in the abdomen and supraclavicular fossae, proximal muscle atrophy, moon face, and multiple hematomas. A lack of a serum cortisol diurnal rhythm with high late-night serum cortisol and undetectable ACTH levels was found (Table 3). The short DST revealed no cortisol suppression (Table 3), and the UFC result was 725 µg/24 h, which exceeded the UNL more than four times. The serum levels of renin, aldosterone, and 24-h urine fractionated metanephrines were within the normal ranges. Computed tomography imaging revealed a left adrenal gland tumor measuring 25 × 26 × 22 mm, with a basal density of 32 HU and a washout rate typical for adenoma (76%).

Table 3 Laboratory Results Before Osilodrostat Therapy – Case 2
Osilodrostat therapy was administered for preoperative management. The initial daily dose was 2 mg/day, increased gradually by 2 mg every day with no serum cortisol response (late night cortisol levels 15.8–18.5 µg/dl) and no AEs of the drug (Table 4). After the daily dose of osilodrostat reached 10 mg, it was escalated by 5 mg every other day, initially with no serum cortisol reduction. The dose was increased to 45 mg daily (with the lowest detected late-night serum cortisol of 9.6 µg/dl) (Table 4).

Table 4 Changes in the Most Important Parameters During Osilodrostat Therapy – Case 2
After a week of administration of 45 mg daily, UFC normalization was achieved. Despite rapid dose escalation, no AEs were observed during the entire therapy period. Potassium levels were normal without any supplementation (the lowest detected serum potassium level was 3.9 mmol/l; all other results were over 4.0 mmol/l) (Table 4). After UFC normalization, left adrenalectomy was performed without complications. Histopathological examination revealed benign adrenal adenoma. Antihypertensive therapy was reduced only to 2.5 mg of nebivolol daily. The patient’s general condition improved significantly. Currently, hydrocortisone replacement therapy is administered at a dose of 15 mg/day.
Discussion
Osilodrostat is a novel potent steroidogenesis inhibitor whose efficacy and safety have been thoroughly analyzed in clinical trials of patients with CD, the most common cause of endogenous hypercortisolism. No clinical trial of osilodrostat therapy in CPA has been performed, as this disease constitutes only 10% of all cases of endogenous hypercortisolism. Moreover, osilodrostat is not approved by the FDA for hypercortisolism conditions other than CD.9 Therefore, data on potential differences in the treatment regimen are lacking.
During the course of already reported trials in CD, osilodrostat doses were escalated slowly, every 2–3 weeks,3,5,6 with an excellent response to quite low doses of the drug.3–6 In the LINC 2 extension study the median average dose was 10.6 mg/day,5 while in the LINC 3 extension study and the LINC 4 study it was 7.4 mg/day and 6.9 mg/day, respectively.4,6 In most cases, a significant decrease of hypercortisolism was reported with the low doses of osilodrostat (4 or 10 mg/day). Moreover, some patients received 1 mg/day or even 1 mg every other day, with a good response.6 Even in rare cases of CD in whom initial short-term etomidate therapy was given at the beginning of osilodrostat therapy, due to highly severe life-threatening symptoms of hypercortisolism, the final effective dose of osilodrostat was much lower than that in our patients with CPA (25 mg/day vs 45 mg/day) and no increase of cortisol level was observed.11
It should be underlined that many cases of adrenal insufficiency during osilodrostat therapy in patients with CD have been reported,3–6,12,13 and – therefore – low initial dose with slow gradual dose escalation is recommended in patients with CD.1,6,13
In the cases presented here, CPA led to severe hypercortisolism, the complications of which constituted contraindications for surgery. Therefore, osilodrostat therapy was introduced as a presurgical treatment. In Case 1, the therapy was started at low doses according to the approved product characteristics.14 Due to the severity of hypertension, which was uncontrolled despite of active antihypertensive therapy, as well as to unstable DM, the doses were increased faster than recommended. Surprisingly, we immediately observed a gradual increase in hypercortisolism, in both serum cortisol levels and the UFC, with simultaneous burst of complications related to both hypercortisolism itself and 11β-hydroxylase inhibition. Life-threatening episodes of hypertensive crisis responded poorly to standard therapies. Severe exaggeration of cardiac insufficiency could probably be related to these episodes as well as to deep hypokalemia, which occurred despite potassium supplementation. Hypokalemia is a typical complication of treatment with 11β-hydroxylase inhibitors due to the accumulation of adrenal hormone precursors. However, Patient 1 required much higher doses of potassium supplementation, both parenteral and oral, than ever described during osilodrostat therapy.3–6,13 The dose of 20 mg/day of osilodrostat was the first one which led to noticeable cortisol reduction and a decrease in systolic blood pressure (SBP) to below 170 mmHg. Surprisingly, instead of the expected deterioration of hypokalemia, parenteral potassium administration could be stopped with an osilodrostat dose of 20 mg/day and oral supplementation was gradually reduced simultaneously with osilodrostat dose escalation. The reason why such severe hypokalemia occurred with low doses of osilodrostat and did not deteriorate further seems complex. One possible reason is the administration of high doses of potassium-saving antihypertensive drugs such as spironolactone and the angiotensin II receptor antagonist telmisartan. Additionally, one can consider other possible mechanisms, such as downregulation of the receptors of deoxycorticosterone (DOC) or other adrenal hormone precursors. However, this hypothesis requires further research and confirmation. Such an improvement of the potassium level during osilodrostat dose escalation was previously demonstrated in a patient with CD.11 Interestingly, in our Patient 2, no potassium supplementation was required during the whole time of osilodrostat therapy, although the doses were increased intensively up to the finally effective dose, which was the same (45 mg/day) as for Patient 1. In Patient 2, no actual response to doses lower than 20 mg/day was observed. UFC normalization was achieved after a week of administration of 45 mg/day, five weeks from the beginning of therapy. Although UFC normalization is not always required in pre-surgical treatment, clinical symptoms significantly improved in our patients only after the UFC upper normal level was achieved.
The present paper is one of only a few reports focused on osilodrostat therapy in CPA, and the only one presenting a different therapy course as compared to patients with CD. No case of CPA resistance to low doses of osilodrostat has been described. It should be underlined that in our report “low doses” of osilodrostat were higher than the average mean doses of osilodrostat used in clinical trials in patients with CD.3–6 Therefore, they should not generally be considered low but only much lower than those which were effective in our patients. Malik and Ben-Shlomo presented a case of CPA treated with osilodrostat, with an immediate decrease in cortisol level at 4 mg/day and adrenal insufficiency symptoms after dose escalation to 8 mg/day.15 Similar to our two cases, their patient was a middle-aged female with normal results of all other adrenal parameters, such as renin, angiotensin, or metanephrine levels. However, a CT scan was not performed (or presented), while magnetic resonance imaging revealed an indeterminate adrenal gland mass without a typical contrast phase/out-of-phase dropout for adenoma.15 Therefore, different morphology of cortisol-secreting adrenal tumor can potentially be considered a reason of the different response to treatment. Tanaka et al performed a multicenter study on the efficacy and safety of osilodrostat in Japanese patients with non-CD Cushing’s syndrome.16 Five patients with CPA were included in the study, and none of them required osilodrostat doses higher than 10 mg/day to achieve UFC normalization. However, most of the patients presented by Tanaka et al were previously treated with metyrapone,16 whereas both of our patients were treatment-naive. Previous metyrapone therapy may be considered as a potential reason of better response to osilodrostat. This hypothesis was confirmed in the quoted study by Tanaka et al, who demonstrated that at week 12 the median percent changes in the mUFC values were higher in patients previously treated with metyrapone (–98.97%) than in treatment-naive cases (–86.65%).16 Detomas et al performed a comparison of efficacy and safety of osilodrostat and metyrapone, with one CPA patients included in a group treated with osilodrostat, however no data on a dose required for a disease control are available separately for this particular patient.8 To the best of our knowledge, no more CPA cases have been described and therefore no further comparison is available.
Higher doses of osilodrostat were administered to a group of seven patients with hypercortisolism due to adrenocortical carcinoma (ACC) presented by Tabarin et al.17 A full control of hypercortisolism was achieved in one patient for each dose of 4, 8, 10, and 20 mg/day, and in three patients treated with 40 mg/day.17 These patients, however received other therapies including mitotane and chemotherapy, which can significantly modify the response to osilodrostat.
Several authors have reported the phenomenon of a partial or total loss of response to osilodrostat.5,16,17 In such cases, a response to treatment was initially achieved and then lost during treatment with the same dose. A further increase in osilodrostat dose usually resulted in the response resumption.5,16,17 Such a situation could not be suspected in either of our cases.
The presented cases provide a novel insight into modalities of treatment with osilodrostat in patients with CPA and demonstrate for the first time that an inverse cortisol response is possible in CPA cases, especially those with a higher CT density of adrenal adenoma. Such a situation should not be considered a contraindication to dose escalation. Conversely, the dose should be increased more intensively so as to achieve the initial efficacy threshold, which was 20 mg/day in both of our patients. The fully efficient dose that allowed UFC normalization was more than twice as high (45 mg/day in both cases). A similar approach should be applied in patients who do not respond to lower doses, such as Patient 2. The safety of osilodrostat therapy is strictly individual and not dose dependent in patients with CPA. Adverse events, including hypokalemia, severe hypertension, and edema, can be of life-threatening severity or may not occur regardless of the dose. Moreover, AEs of high severity may decrease with osilodrostat dose escalation. Our study demonstrated that osilodrostat is efficient and can be used in patients with CPA as a pre-surgical therapy if surgery is contraindicated due to hypercortisolism complications.
Our study presented two cases of CPA treated with osilodrostat, and a small size of our group is the main limitation of this report. Future research is required to confirm our observations.
Conclusion
In some patients with CPA, the doses of osilodrostat required for disease control can be much higher than those previously reported. Acceleration of the dose increase can be fast, and the risk of overdosing, adrenal insufficiency, and later necessity of dose reduction seem to be much lower than it could be expected. Low initial doses (<20 mg/day in our study) can be entirely ineffective or can even cause exacerbation of hypercortisolism, whereas high doses (45 mg/day in the present study) are efficient in pre-surgery UFC normalization. AEs associated with osilodrostat can be rapid, with severe hypokalemia despite active potassium supplementation, or may not occur even if high doses of osilodrostat are applied. Therefore, close monitoring for potential AEs is necessary.
Acknowledgments
The abstract included some parts of this paper was presented at the European Congress of Endocrinology ECE2023 as a rapid communication. The abstract was published in the Endocrine Abstracts Vol. 90 [https://www.endocrine-abstracts.org/ea/0090/].
Funding
The publication of this report was financially supported by the statutory funds of the Polish Mother’s Memorial Hospital – Research Institute, Lodz, Poland.
Disclosure
Professor Przemysław Witek reports personal fees from Investigator in the clinical trials paid by Novartis and Recordati Rare Diseases, outside the submitted work; lectures fees from Recordati Rare Diseases, Strongbridge, IPSEN. The authors report no other conflicts of interest in this work.
References
1. Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol. 2021;9(12):847–875. doi:10.1016/S2213-8587(21)00235-7
2. Pivonello R, Isidori AM, De Martino MC, et al. Complications of Cushing’s syndrome: state of the art. Lancet Diabetes Endocrinol. 2016;4(7):611–629. doi:10.1016/S2213-8587(16)00086-3
3. Pivonello R, Fleseriu M, Newell-Price J, et al. Efficacy and safety of osilodrostat in patients with Cushing’s disease (LINC 3): a multicentre Phase III study with a double-blind, randomised withdrawal phase. Lancet Diabetes Endocrinol. 2020;8(9):48–761. doi:10.1016/S2213-8587(20)30240-0
4. Fleseriu M, Newell-Price J, Pivonello R, et al. Long-term outcomes of osilodrostat in Cushing’s disease: LINC 3 study extension. Eur J Endocrinol. 2022;187(4):531–541. doi:10.1530/EJE-22-0317
5. Fleseriu M, Biller BMK, Bertherat J, et al. Long-term efficacy and safety of osilodrostat in Cushing’s disease: final results from a Phase II study with an optional extension phase (LINC 2). Pituitary. 2022;25(6):959–970. doi:10.1007/s11102-022-01280-6
6. Gadelha M, Bex M, Feelders RA, et al. Randomized trial of osilodrostat for the treatment of Cushing disease. J Clin Endocrinol Metab. 2022;107(7):e2882–e2895. doi:10.1210/clinem/dgac178
7. Daniel E, Aylwin S, Mustafa O, et al. Effectiveness of metyrapone in treating cushing’s syndrome: a retrospective multicenter study in 195 patients. J Clin Endocrinol Metab. 2015;100(11):4146–4154. doi:10.1210/jc.2015-2616
8. Detomas M, Altieri B, Deutschbein T, et al. Metyrapone versus osilodrostat in the short-term therapy of endogenous cushing’s syndrome: results from a single center cohort study. Front Endocrinol. 2022;13:903545. doi:10.3389/fendo.2022.903545
9. U.S. food and drug administration home page. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-adults-cushings-disease. Accessed March 22, 2023.
10. Agency for health technology assessment and tariff system home page. Available from: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&cad=rja&uact=8&ved=2ahUKEwj6ypGbsfT9AhUMzYsKHTgAD2EQFnoECA8QAQ&url=https%3A%2F%2Fbipold.aotm.gov.pl%2Fassets%2Ffiles%2Fwykaz_tli%2FRAPORTY%2F2020_010.pdf&usg=AOvVaw3P2Q85gwi3JcxKkW3uxfOb. Accessed March 22, 2022.
11. Dzialach L, Sobolewska J, Respondek W, et al. Cushing’s syndrome: a combined treatment with etomidate and osilodrostat in severe life-threatening hypercortisolemia. Hormones. 2022;21(4):735–742. doi:10.1007/s42000-022-00397-4
12. Ekladios C, Khoury J, Mehr S, et al. Osilodrostat-induced adrenal insufficiency in a patient with Cushing’s disease. Clin Case Rep. 2022;10(11):e6607. doi:10.1002/ccr3.6607
13. Fleseriu M, Biller BMK. Treatment of Cushing’s syndrome with osilodrostat: practical applications of recent studies with case examples. Pituitary. 2022;25(6):795–809. doi:10.1007/s11102-022-01268-2
14. Summary of product characteristics. Available from: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&ved=2ahUKEwim1_KdsvT9AhVq-ioKHUZKAc4QFnoECA4QAQ&url=https%3A%2F%2Fwww.ema.europa.eu%2Fen%2Fdocuments%2Fproduct-information%2Fisturisa-epar-product-information_pl.pdf&usg=AOvVaw0S8nayCTdqNh1LsEcXVLEu. Accessed March 24, 2023.
15. Malik RB, Ben-Shlomo A. Adrenal cushing’s syndrome treated with preoperative osilodrostat and adrenalectomy. AACE Clin Case Rep. 2022;8(6):267–270. doi:10.1016/j.aace.2022.10.001
16. Tanaka T, Satoh F, Ujihara M, et al. A multicenter, Phase 2 study to evaluate the efficacy and safety of osilodrostat, a new 11β-hydroxylase inhibitor, in Japanese patients with endogenous Cushing’s syndrome other than Cushing’s disease. Endocr J. 2020;67(8):841–852. doi:10.1507/endocrj.EJ19-0617
17. Tabarin A, Haissaguerre M, Lassole H, et al. Efficacy and tolerance of osilodrostat in patients with Cushing’s syndrome due to adrenocortical carcinomas. Eur J Endocrinol. 2022;186(2):K1–K4. doi:10.1530/EJE-21-1008
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License. By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License. By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
-
1
-
-
From all the news items I share, and the patients I talk to, it seems like not a lot of advancement is being made over the years. I hear about more people thinking that they have Cushing's after reading about some of the symptoms but it still seems hard for them to get to good doctors, to get diagnosed

Hopefully, next year will be better news!
-
1
-
-
Even though I posted this in 2022 - the original website is still being redone. Probably it will be next year, too! LOL
-
1
-
-
Abstract
Disclosure: C.M. Godar: None. E.B. Noble: None. N.O. Vietor: None. T.S. Knee: None.
Background: Cushing’s syndrome may rarely present as an emergency known as Florid Cushing’s Syndrome. Patients can exhibit severe hyperglycemia, hypertension, hypokalemia, infections, and hypercoagulability. Cushing’s syndrome is a rare disease, and the constellation of clinical features can be overlooked if clinicians are not aware of the manifestations of hypercortisolism. We present the case of a patient with Cushing’s syndrome that went unrecognized with life-threatening sequelae.
Case presentation: A 52-year-old woman with well-controlled type 2 diabetes and hypertension was admitted to the hospital for severe left lower extremity cellulitis. Prior to hospitalization she had noted rapid weight gain, fatigue, weakness, mental clouding, and moodiness. She was admitted for antibiotics and surgical debridement. The infection persisted despite broad spectrum antibiotics, multiple surgical debridements, and skin grafting. She became bacteremic, and extremity amputation was considered. She additionally developed hypertensive emergency, refractory hypokalemia, and hyperglycemia to 396 mg/dL. Exam was notable for facial plethora, supraclavicular fullness, dorsocervical fat pad, and violaceous abdominal striae. Cushing’s Syndrome was suspected, and labs revealed a significantly elevated random serum cortisol of 60.5mcg/dL (Ref 6.2-19.4), significantly elevated 24H urine cortisol of 2157mcg/24H (Ref 0-50), and ACTH elevated to 81.8pg/mL (Ref 7.2-63.3) that confirmed Cushing’s Disease. MRI sella and octreotide scans did not localize a lesion. Inpatient therapy included multiple antihypertensive agents, insulin drip, aggressive potassium repletion, and initiation of ketoconazole to reduce cortisol levels. Ketoconazole was maximally dosed and she underwent surgical exploration and removal of a small pituitary microadenoma. Following surgery, she developed transient adrenal insufficiency requiring hydrocortisone and she no longer required antihypertensives, insulin, or potassium therapy. Follow up 7 years later has revealed no recurrence of Cushing’s Disease.
Discussion: Cushing’s Syndrome may present with a variety of clinical features and rarely may present as a medical emergency. Delay in diagnosis can lead to Florid Cushing’s Syndrome which carries high risk for morbidity and mortality. This case illustrates the need for clinician awareness of the features of Cushing’s Syndrome: hypertension, hyperglycemia, rapid weight gain, cushingoid exam features, hypokalemia, hirsutism, virilization, infection, and/or hypercoagulable state. Severe hypercortisolism was responsible for this patient’s refractory infection, and if not controlled, she likely would have endured a lower extremity amputation. Rapid detection with elevated random serum and/or urine cortisol and treatment with a cortisol-lowering agent is critical and lifesaving.
Presentation: Thursday, June 15, 2023
This content is only available as a PDF.-
1
-
-
Two years later
“My Dream Day“…
I’d wake up refreshed and really awake at about 7:00AM and
take the dog out(no more dog) for a brisk run. Maybe do a Silver Sneakers online workout unless it's a water aerobics day.Get home about 8:00AM and start on my website work.
Later in the morning, I’d get some bills paid – and there would be enough money to do so!
After lunch,
out with the dog again, then practice the piano some, practice my balalaika and tenor recorder son, read a bit, finish up the website work, teach a few piano students, work on my church job, then dinner.After dinner, check email,
out with the dog, maybe handbell or choir practice, a bit of TV, then bed about 10PMNothing fancy but NO NAPS. Work would be getting done, time for hobbies,
the dog, 3 healthy meals.Just a normal life that so many take for granted. Or, do they?
I would love to have enough money to retire from some of those jobs, to travel more, to cruise more...
-
Highlights
- Aim to identify independent risk factors for postoperative delirium after pituitary adenoma surgery.
- Select matched subjects by Propensity Score Matching to reduce potential biases caused by variables.
- Enhance preoperative communication to minimize the occurrence of delirium, for patients at high risk of postoperative delirium.
- Minimize surgery duration and general anesthesia, optimize perioperative sedation regimen.
- Reducing unnecessary or excessive protective physical restraints.
Abstract
Objectives
The primary aim of this study is to explore the factors associated with delirium incidence in postoperative patients who have undergone endoscopic transsphenoidal approach surgery for pituitary adenoma.
Methods
The study population included patients admitted to Tianjin Huanhu Hospital's Skull Base Endoscopy Center from January to December 2022, selected through a retrospective cohort study design. The presence of perioperative delirium was evaluated using the 4 'A's Test (4AT) scale, and the final diagnosis of delirium was determined by clinicians. Statistical analysis included Propensity Score Matching (PSM), χ2 Test, and Binary Logistic Regression.
Results
A total of 213 patients were included in this study, and the incidence of delirium was found to be 29.58 % (63/213). Among them, 126 patients were selected using PSM (delirium:non-delirium = 1:1), ensuring age, gender, and pathology were matched. According to the results of univariate analysis conducted on multiple variables, The binary logistic regression indicated that a history of alcoholism (OR = 6.89, [1.60–29.68], P = 0.010), preoperative optic nerve compression symptoms (OR = 4.30, [1.46–12.65], P = 0.008), operation time ≥3 h (OR = 5.50, [2.01–15.06], P = 0.001), benzodiazepines for sedation (OR = 3.94, [1.40–11.13], P = 0.010), sleep disorder (OR = 3.86, [1.40–10.66], P = 0.009), and physical restraint (OR = 4.53, [1.64–12.53], P = 0.004) as independent risk factors for postoperative delirium following pituitary adenoma surgery.
Conclusions
For pituitary adenoma patients with a history of alcoholism and presenting symptoms of optic nerve compression, as well as an operation time ≥3 h, enhancing communication between healthcare providers and patients, improving perioperative sleep quality, and reducing physical restraint may help decrease the incidence of postoperative delirium.
Introduction
In clinical practice, patients admitted to the intensive care unit (ICU) during the postoperative period after endoscopic transsphenoidal tumorectomy of pituitary adenoma often experience episodes of delirium. According to a recent retrospective analysis conducted at a single center, the incidence of postoperative delirium among these patients was found to be 10.34 % (n = 360) [1]. Delirium is a common complication following neurosurgery, characterized by acute distraction, confusion in thinking, sleep disorders, and cognitive decline. The incidence of delirium in admitted patients after neurosurgery has been reported to be 19 %, with a range of 12 % to 26 % depending on clinical features and the methods used for delirium assessment [2], [3], [4]. The incidence of postoperative delirium varied across different types of neurosurgical diseases, as reported in a meta-analysis [2]. Specifically, the incidences were 8.0 % for patients with neurological tumors, 20 % for those undergoing functional neurosurgery, 24.0 % for microvascular decompression patients, 19.0 % for traumatic brain injury patients, 42.0 % for neurovascular patients, and 17.0 % for the mixed population undergoing neurosurgery procedures. Furthermore, the incidence rates of delirium in intensive care units (ICUs), general wards, or both combined were found to be 24.0 %, 17 %, and 18 %, respectively.
The aforementioned issue not only leads to prolonged hospital stays and increased healthcare costs, but also exerts a significant impact on patient consciousness and cognitive function. Therefore, early and accurate identification of delirium in post-neurosurgical patients is crucial. However, due to frequent co-occurrence with primary brain injury, related complications can also lead to cognitive impairment or even decreased levels of consciousness, posing challenges for timely and precise identification of delirium. Currently, the primary focus lies in the prevention of delirium within the neurosurgical ICU setting. Early identification and comprehensive pre-surgical assessment are positively significant measures for preventing postoperative delirium occurrence [5], [6]. In this study, a retrospective cohort design was employed to collect pertinent data and statistically analyze the incidence of delirium, as well as its associated influencing factors, among patients admitted to the neurosurgical ICU for pituitary adenoma treatment. And now it is reported as follows.
Section snippets
Patient selection
A retrospective cohort study design was employed to select 213 pituitary adenomas admitted to the Skull Base and Endoscopy Center of Tianjin Huanhu Hospital between January 2022 and December 2022 as the subjects for investigation, with a review of their medical records. The mean age was (50.03 ± 15.72) years, ranging from 20–79 years old (Fig. 1). Informed consent was obtained from all patients or their families, ensuring compliance with the requirements stated in the Declaration of Helsinki.
Inclusion criteria
a.
Propensity score matching
The present study enrolled a total of 213 patients with pituitary tumors, among whom 63 exhibited symptoms related to delirium while the remaining 150 did not. Consequently, the incidence rate of delirium was determined to be 29.58 % in this cohort of patients admitted to the intensive care unit following pituitary tumor surgery. The univariate analysis revealed no significant differences in age (≥65y old, 23.8 % vs. 23.3 %, P = 0.940) and gender (male, 49.2 % vs. 56.7 %, P = 0.318) between the
Background of perioperative delirium in transsphenoidal endoscopic pituitary adenoma surgery
The pituitary gland is situated within the sella turcica and comprises two distinct components. The anterior pituitary, known as the adenohypophysis, functions as an endocrine organ responsible for secreting growth hormone, prolactin, adrenocorticotropic hormone, thyrotropin, follicle-stimulating hormone and luteinizing hormone. On the other hand, the posterior pituitary, referred to as the neurohypophysis, serves as a direct extension of the hypothalamus and acts as a storage site for
Conclusions
To enhance the evaluation of postoperative patients at risk of delirium, it is anticipated that optimizing doctor-nurse-patient communication and minimizing unnecessary and indiscriminate protective measures will mitigate the incidence of delirium following pituitary tumor surgery. This study is a single-center prospective study conducted at our institution, which has several inherent limitations. A large-scale multicenter prospective study is anticipated to further investigate the associated
Limitations
There are multiple factors that influence the occurrence of delirium following neurosurgery. This retrospective study solely focused on analyzing and comparing general patient data, medical history, and potential perioperative factors contributing to delirium, without considering any other known or unknown variables in this analysis. The pituitary gland functions as a neuroendocrine organ involved in the regulation of neuroendocrine processes. Changes in hormone levels following surgery for
Funding
All authors affirm that this study was conducted without any fund support from external organizations.
CRediT authorship contribution statement
Shusheng Zhang: Writing – original draft, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Yanan Chen: Writing – original draft, Investigation, Data curation. Xiudong Wang: Validation, Supervision, Project administration, Methodology, Conceptualization. Jun Liu: Software, Formal analysis, Data curation. Yueda Chen: Validation, Supervision, Methodology, Investigation. Guobin Zhang: Writing – review & editing, Validation, Supervision, Methodology, Conceptualization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References (21)
-
G. Sousa et al.
Postoperative delirium in patients with history of alcohol abuse
Rev Esp Anestesiol Reanim
(2017) -
M. Terzaghi et al.
Sleep disorders and acute nocturnal delirium in the elderly: a comorbidity not to be overlooked
Eur J Intern Med
(2014) -
S. Lee et al.
Opioid and benzodiazepine use in the emergency department and the recognition of delirium within the first 24 hours of hospitalization
J Psychosom Res
(2022) -
A.J. Slooter et al.
Delirium in critically ill patients
Handb Clin Neurol
(2017) -
E. Wang et al.
Effect of perioperative benzodiazepine use on intraoperative awareness and postoperative delirium: a systematic review and meta-analysis of randomized controlled trials and observational studies
Br J Anaesth
(2023) -
E. Rollo et al.
Physical restraint precipitates delirium in stroke patients
J Neurol Sci
(2021) -
H. Chen et al.
The incidence and predictors of postoperative delirium after brain tumor resection in adults: A cross-sectional survey
World Neurosurg
(2020) -
J. Liu et al.
Identifying hormones and other perioperative risk factors for postoperative delirium after endoscope-assisted transsphenoidal pituitary adenoma resection: A retrospective, matched cohort study
Brain Behav
(2023) -
P.R. Kappen et al.
Delirium in neurosurgery: a systematic review and meta-analysis
Neurosurg Rev
(2022) -
J. Wang et al.
Risk factors for the incidence of delirium in cerebrovascular patients in a Neurosurgery Intensive Care Unit: A prospective study
J Clin Nurs
(2018)
There are more references available in the full text version of this article. -
Happy Birthday, Harvey!
-
1
-
-

It's Sunday again, so this is another semi-religious post so feel free to skip it
I'm sure that many would think that Abide With Me is a pretty strange choice for my all-time favorite hymn, especially since it often shows up at funerals and memorial services.
My dad was a Congregational (now United Church of Christ) minister so I was pretty regular in church attendance in my younger years.
Some Sunday evenings, he would preach on a circuit and I'd go with him to some of these tiny churches. The people there, mostly older folks, liked the old hymns best - Fanny Crosby and so on.
So, some of my "favorite hymns" are those that I sang when I was out with my Dad. Fond memories from long ago.
In 1986 I was finally diagnosed with Cushing's after struggling with doctors and trying to get them to test for about 5 years. I was going to go into the NIH (National Institutes of Health) in Bethesda, MD for final testing and then-experimental pituitary surgery.
I was terrified and sure that I wouldn't survive the surgery.
Somehow, I found a 3-cassette tape set of Reader's Digest Hymns and Songs of Inspiration and ordered that. The set came just before I went to NIH and I had it with me.
At NIH I set up a daily "routine" of sorts and listening to these tapes was a very important part of my day and helped me get through the ordeal of more testing, surgery, post-op and more.
When I had my kidney cancer surgery, those tapes were long broken and irreplaceable, but I had replaced all the songs - this time on my iPod.
Abide With Me was on this original tape set and it remains a favorite to this day. Whenever we have an opportunity in church to pick a favorite, my hand always shoots up and I request page 700. When someone in one of my handbell groups moves away, we always sign a hymnbook and give it to them. I sign page 700.
I think that many people would probably think that this hymn is depressing. Maybe it is but to me it signifies times in my life when I thought I might die and I was so comforted by the sentiments here.
This hymn is often associated with funeral services and has given hope and comfort to so many over the years - me included.
If you abide in Me, and My words abide in you, you will ask what you desire, and it shall be done for you.
~John 15:7
Abide With Me
Words: Henry F. Lyte, 1847.
Music: Eventide, William H. Monk, 1861. Mrs. Monk described the setting:
This tune was written at a time of great sorrow—when together we watched, as we did daily, the glories of the setting sun. As the last golden ray faded, he took some paper and penciled that tune which has gone all over the earth.
Lyte was inspired to write this hymn as he was dying of tuberculosis; he finished it the Sunday he gave his farewell sermon in the parish he served so many years. The next day, he left for Italy to regain his health. He didn’t make it, though—he died in Nice, France, three weeks after writing these words. Here is an excerpt from his farewell sermon:
O brethren, I stand here among you today, as alive from the dead, if I may hope to impress it upon you, and induce you to prepare for that solemn hour which must come to all, by a timely acquaintance with the death of Christ.
For over a century, the bells of his church at All Saints in Lower Brixham, Devonshire, have rung out “Abide with Me” daily. The hymn was sung at the wedding of King George VI, at the wedding of his daughter, the future Queen Elizabeth II, and at the funeral of Nobel peace prize winner Mother Teresa of Calcutta in1997.
Abide with me; fast falls the eventide;
The darkness deepens; Lord with me abide.
When other helpers fail and comforts flee,
Help of the helpless, O abide with me.
Swift to its close ebbs out life’s little day;
Earth’s joys grow dim; its glories pass away;
Change and decay in all around I see;
O Thou who changest not, abide with me.
Not a brief glance I beg, a passing word;
But as Thou dwell’st with Thy disciples, Lord,
Familiar, condescending, patient, free.
Come not to sojourn, but abide with me.
Come not in terrors, as the King of kings,
But kind and good, with healing in Thy wings,
Tears for all woes, a heart for every plea—
Come, Friend of sinners, and thus bide with me.
Thou on my head in early youth didst smile;
And, though rebellious and perverse meanwhile,
Thou hast not left me, oft as I left Thee,
On to the close, O Lord, abide with me.
I need Thy presence every passing hour.
What but Thy grace can foil the tempter’s power?
Who, like Thyself, my guide and stay can be?
Through cloud and sunshine, Lord, abide with me.
I fear no foe, with Thee at hand to bless;
Ills have no weight, and tears no bitterness.
Where is death’s sting? Where, grave, thy victory?
I triumph still, if Thou abide with me.
Hold Thou Thy cross before my closing eyes;
Shine through the gloom and point me to the skies.
Heaven’s morning breaks, and earth’s vain shadows flee;
In life, in death, O Lord, abide with me.
http://cushieblog.files.wordpress.com/2012/04/maryo-butterfly-script1.gif
-
1
-
-
-
-
Abstract
Avascular necrosis (AVN), also called osteonecrosis, stems from blood supply interruption to the bone and is often idiopathic. It has risk factors like trauma, alcohol, and corticosteroids. AVN in the talus (AVNT) is less common than in the femoral head. Most cases of talar osteonecrosis are associated with trauma, while a smaller proportion is linked to systemic conditions such as sickle cell disease or prolonged prednisone use. Glucocorticoids are a key nontraumatic cause. We report a middle-aged woman with Cushing’s syndrome symptoms, such as hypertension and moon face, since her youth. A few years ago, she experienced pain and swelling in her ankle, which was diagnosed as atraumatic AVNT and treated with hindfoot fusion. Years later, she was diagnosed with Cushing’s disease caused by an adrenocorticotropic hormone (ACTH)-producing pituitary adenoma in laboratory tests and imaging, which was resected in 2020. She experienced significant weight loss, and her Cushing’s syndrome symptoms were relieved after tumor resection. Mechanisms behind AVN in hypercortisolism involve fat cell hypertrophy, fat embolization, osteocyte apoptosis, and glucocorticoid-induced hypertension. Traditional X-rays may miss early AVN changes; MRI is preferred for early detection. Although there are some cases of femoral AVN caused by endogenous hypercortisolism in the literature, as far as we know, this is the first case of AVNT due to Cushing’s disease. AVNT treatment includes conservative approaches, hindfoot fusion, and core decompression. Cushing’s disease is a rare cause of AVNT, and a multidisciplinary approach aids in the rapid diagnosis of elusive symptoms.
Introduction
Avascular necrosis (AVN), also known as osteonecrosis, is a condition arising from the temporary interruption or permanent cessation of blood supply to a bone, leading to tissue necrosis or its demise. While AVN is frequently idiopathic, certain established risk factors are known including trauma, alcohol abuse, and the use of exogenous corticosteroids [1]. While not as prevalent as in the femoral head, AVN of the talus (AVNT) in the ankle presents a painful and disabling issue for patients and poses a challenging dilemma for orthopedic surgeons [2]. About 75% of cases of talar osteonecrosis stem from traumatic injuries, while approximately 25% of nontraumatic instances are typically associated with systemic conditions such as sickle cell disease or prolonged use of prednisone, which impede blood flow. [3]
The use of glucocorticoids is one of the most important non-traumatic causes of AVN. Nevertheless, there are some case reports where AVN in the femoral head is reported as a manifestation of endogenous hypercortisolism, particularly associated with Cushing's syndrome [4-12].
In this article, we describe the case of a middle-aged woman who was diagnosed with idiopathic severe progressive AVNT for two years. She had retrogradely diagnosed masked symptoms of Cushing’s disease since her youth, but the diagnosis was confirmed after undergoing ankle arthrodesis.
Case Presentation
A 43-year-old woman visited our office in June 2018 with a complaint of severe pain and swelling in her left ankle, which had persisted for the past two years. She had hypertension since her youth and blurry vision since 2013 but had no other significant medical or family history. She was also diagnosed with major depressive disorder (MDD) in 2015 when she lost her husband. She had no history of smoking, alcohol consumption, or addiction. She had not experienced any significant trauma during this period and sought consultations from various specialties, including neurology, psychology, internal medicine, nephrology, rheumatology, and orthopedics. She had received a platelet-rich plasma (PRP) injection in the ankle, but it did not improve her symptoms. Despite undergoing various diagnostic workups, no precise diagnosis had been established.
Back in 2013, she remembers suddenly experiencing blurry vision in her right eye. This condition underwent a misdiagnosis, which almost led to a loss of vision. She had been struggling with her eye problems until her last visit, during which she received intravitreal bevacizumab injections. Additionally, she previously had iron deficiency anemia, which was treated with ferrous sulfate before 2018.
In our first visit, during the physical examination, the pain was localized in the ankle mortise with some posterolateral pain along the course of the peroneal tendons posterior to the fibula. Based on the physical examination and available ankle radiographs, we diagnosed AVNT. The patient subsequently underwent ankle arthroscopy through the standard anterior portals, the joint was cleaned, the synovium was shaved, and a small incision was conducted for peroneal assessment; this procedure revealed a subchondral collapse and extensive necrosis in the talus. Following the procedure, she experienced a partial improvement in her symptoms. However, six months later, she returned with a recurrence of symptoms (Figure 1). Upon further inquiry, she mentioned that her symptoms had recurred a month ago when she was dancing at a family party. Radiographs showed a stress fracture in her fibula and extensive AVNT. This diagnosis was confirmed through a CT scan, MRI, and bone scan (Figure 2).

Figure 1: Ankle X-ray six months after arthroscopy
Pain had reduced for four months, then pain increased with activity and disabled her after a night of dancing. Subchondral fracture and fibular stress fracture are evident (A and B, respectively).
-bone-scan")
Figure 2: MRI, CT scan, and technetium-99m (Tc-99m) bone scan
Coronal MRI confirmed avascular necrosis of the talar dome with subchondral fracture (A and B, respectively). CT scan (C) and Tc-99 bone scan (D) images also revealed the pathologies.
In the second visit after arthroscopy, upon confirmation of a fibular stress fracture and significant subchondral collapse, and following a discussion of the next available options with the patient, the second procedure was performed as an ankle arthrodesis with an anterior approach. A 6 cm longitudinal incision was made anteriorly, and through the plane between the tibialis anterior and extensor hallucis longus, the ankle joint was accessed. Joint preparation was done with an osteotome, ensuring a bleeding surface on both sides. Then, manual compression with provisional pin fixation in the corrective position was performed. The fusion was planned at less than 5 degrees of valgus, 10 degrees of external rotation, and approximately 10 degrees of plantar flexion, suitable for the high-heeled shoes that she was using in her daily living activities. After confirming fluoroscopy in two planes, final 6.5 mm cannulated cancellous screws were used, and fixation was augmented with an anterior molded 4.5 mm narrow dynamic compression plate (DCP), according to our previously published anterior ankle fusion technique [13]. The foot was placed in a splint for 10 days, after which stitches were removed, and a cast was applied for four weeks. Then, walking with gradual, as-tolerated weight-bearing was initiated (Figure 3). Three months after surgery, she was pain-free, and by the sixth month, she could walk without any boot or brace, only using high-heeled shoes.

Figure 3: Post-operative radiographies
Six months after the ankle surgery, a huge osteonecrosis and fibular stress fracture were managed with an acceptable, painless ankle fusion (not solid) despite the remaining necrosis (A and B, respectively). In 2024, four years after the tumor resection, complete healing of talus necrosis and solid fusion were achieved (C and D, respectively).
In 2020, two years after her ankle surgery, she was referred to an endocrinologist due to excessive weight gain and hirsutism. The biochemical assessment revealed the following: cortisol (8 AM) (chemiluminescence immunoassay (CLIA)) was 96 µg/dl (normal range: 4.82 - 19.5 µg/dl), and it was 22.1 µg/dl after overnight dexamethasone (normal range: < 1.8 µg/dl). Adrenocorticotropic hormone (ACTH) (CLIA) was 44.4 pg/ml (normal range: 7.2-63.3 pg/ml), and cortisol measured 5.7 µg/dl after the 48-hour low-dose dexamethasone suppression test (normal < 5 µg/dl). The results, along with symptoms (Table 1), are documented in the laboratory tests (Table 2). She was diagnosed with Cushing’s syndrome, which was subsequently confirmed as Cushing's disease due to an ACTH-producing pituitary adenoma observed in the MRI and Brain CT (Figure 4).
Sign/symptom Severity Weight Gain Severe Hirsutism Severe Hypertension Severe Easy bruising Severe Depression Severe Moon face Moderate (masked with makeup) Lethargy Moderate Headache Moderate Peripheral edema _ Buffalo hump _ Myopathy _ Acne _ Purple striae _ Table 1: Cushing's disease symptoms and signs
The hyphens in the table indicate that the patient does not have those symptoms or signs.
Laboratory test Result Reference range Cortisol (8 AM) (CLIA) 96 µg/dl 4.82-19.5 µg/dl Cortisol (8 AM) (after overnight dexamethasone) (CLIA) 22.1 µg/dl <1.8 µg/dl ACTH (CLIA) 44.4 pg/ml 7.2-63.3 pg/ml Cortisol after 48 hours of LDDST (CLIA) 5.7 µg/dl < 5 µg/dl Table 2: Laboratory tests
CLIA: chemiluminescence immunoassay; ACTH: adrenocorticotropic hormone; LDDST: low-dose dexamethasone suppression test

Figure 4: Brain MRI
Finally, a pituitary adenoma was diagnosed using a Brain MRI as the cause of Cushing’s disease symptoms (A and B).
Finally, she underwent a tumor resection and had a dramatic response after treatment (30 kg weight loss). She revealed that she had Cushing’s syndrome symptoms since she was young. These symptoms included a puffy face, which she covered with makeup, high blood pressure, and hirsutism. In January 2024, four years after her brain surgery, during our last visit, her symptoms had significantly improved. She reported no problems with her ankle, and talus necrosis was completely healed, with a solid fusion achieved in radiographs (Figure 3).
Discussion
As far as we are aware, this case presentation represents the first instance of AVNT attributed to Cushing’s disease in the existing literature. Nevertheless, some individuals with endogenous Cushing's syndrome have been reported to experience AVN of the femoral head [4-12].
Cushing's syndrome is an uncommon endocrine condition marked by manifestations of hypercortisolism. The predominant cause is often an adenoma in the anterior pituitary gland that produces ACTH, referred to as Cushing's disease [14]. The presentation of Cushing's syndrome can vary significantly in both adults and children, influenced by the extent and duration of hypercortisolemia. However, the typical signs and symptoms of Cushing's syndrome are widely known [15]. Although some individuals may perceive these alterations as normal and physiological, the disease can go unnoticed for an extended period, as in our case, in which it remained undiagnosed for more than 20 years.
However, it is known that steroid use is a significant contributing factor to the occurrence of bone osteonecrosis, accounting for up to 40% of non-traumatic instances of AVN [16]. The mechanisms leading to AVN due to either endogenous hypercortisolism or excess exogenous glucocorticoids are not completely understood. There are just some hypotheses that suggest that the hypertrophy of fat cells, embolization of fat, and osteocytes' apoptosis result in impaired blood flow in the bone, ultimately causing ischemic tissue necrosis [17]. An alternative proposed theory suggests that elevated levels of glucocorticoids may cause insulin resistance and subsequently contribute to AVN [18].
Traditional X-rays often fail to detect the initial changes of AVN (as observed in our case). MRI stands as the preferred method for identifying AVN in its early phases, providing an opportunity for timely therapeutic interventions [19,20].
In an analysis of 321 cases of AVNT, the predominant treatment modalities included conservative therapies (n = 104), hindfoot fusion (n = 62), and core decompression (n = 85) [21]. These approaches reflect the primary methods employed in contemporary clinical practice for addressing AVNT.
After all, we confirmed the AVNT diagnosis using MRI and bone scan and managed it with hindfoot fusion. Subsequently, the underlying issue, endogenous hypercortisolism due to an ACTH-producing pituitary adenoma, was identified and treated through resection of the tumor (Figure 5).

Figure 5: Case report timeline
* Avascular necrosis in the talus
Conclusions
Cushing’s syndrome is a rare endocrine disorder characterized by excessive cortisol levels, commonly caused by an ACTH-producing adenoma in the pituitary gland, known as Cushing’s disease. Cushing’s disease may be one of the rare causes of AVNT. To the best of our knowledge, this is the first instance of AVNT due to Cushing’s disease described in the literature. Since atraumatic AVNT is rare in itself, a multidisciplinary approach can lead us to a more rapid and proper diagnosis, as each symptom may be masked or considered rare within its subspecialty field.
References
- Chang CC, Greenspan A, Gershwin ME: Osteonecrosis: current perspectives on pathogenesis and treatment. Semin Arthritis Rheum. 1993, 23:47-69. 10.1016/s0049-0172(05)80026-5
- Zhang H, Fletcher AN, Scott DJ, Nunley J: Avascular osteonecrosis of the talus: current treatment strategies. Foot Ankle Int. 2022, 43:291-302. 10.1177/10711007211051013
- Parekh SG, Kadakia RJ: Avascular necrosis of the talus. J Am Acad Orthop Surg. 2021, 29:e267-78. 10.5435/JAAOS-D-20-00418
- Belmahi N, Boujraf S, Larwanou MM, El Ouahabi H: Avascular necrosis of the femoral head: an exceptional complication of Cushing's disease. Ann Afr Med. 2018, 17:225-7. 10.4103/aam.aam_75_17
- Salazar D, Esteves C, Ferreira MJ, Pedro J, Pimenta T, Portugal R, Carvalho 😧 Avascular femoral necrosis as part of Cushing syndrome presentation: a case report. J Med Case Rep. 2021, 15:287. 10.1186/s13256-021-02882-7
- Alaya Z, Braham M, Bouajina E: Aseptic femur head necrosis revealing Cushing's disease: a rare presentation. J Clin Surg Res. 2020, 1:10.31579/2768-2757/002
- Phillips KA, Nance EP Jr, Rodriguez RM, Kaye JJ: Avascular necrosis of bone: a manifestation of Cushing's disease. South Med J. 1986, 79:825-9. 10.1097/00007611-198607000-00011
- Koch CA, Tsigos C, Patronas NJ, Papanicolaou DA: Cushing's disease presenting with avascular necrosis of the hip: an orthopedic emergency. J Clin Endocrinol Metab. 1999, 84:3010-2. 10.1210/jcem.84.9.5992
- Modroño N, Torán CE, Pavón I, Benza ME, Guijarro G, Navea 😄 Cushinǵs syndrome and avascular hip necrosis: presentation of two patients [Article in Spanish]. Rev Clin Esp (Barc). 2014, 214:e93-6. 10.1016/j.rce.2014.05.003
- Camporro F, Bulacio E, Gutiérrez Magaldi I: Bilateral osteonecrosis of the hip secondary to endogenous Cushing's syndrome due to a recently-diagnosed carcinoid tumour of the lung [Article in Spanish]. Med Clin (Barc). 2016, 147:228. 10.1016/j.medcli.2016.03.042
- Ha JS, Cho HM, Lee HJ, Kim SD: Bilateral avascular necrosis of the femoral head in a patient with asymptomatic adrenal incidentaloma. Hip Pelvis. 2019, 31:120-3. 10.5371/hp.2019.31.2.120
- Anand A, Jha CK, Singh PK, Sinha U, Ganesh A, Bhadani PP: Avascular necrosis of femur as a complication of Cushing's syndrome due to adrenocortical carcinoma. Am Surg. 2023, 89:2701-4. 10.1177/00031348221129510
- Gharehdaghi M, Rahimi H, Mousavian A: Anterior ankle arthrodesis with molded plate: technique and outcomes. Arch Bone Jt Surg. 2014, 2:203-9.
- Lindholm J, Juul S, Jørgensen JO, et al.: Incidence and late prognosis of cushing's syndrome: a population-based study. J Clin Endocrinol Metab. 2001, 86:117-23. 10.1210/jcem.86.1.7093
- Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, Montori VM: The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008, 93:1526-40. 10.1210/jc.2008-0125
- Konarski W, Poboży T, Konarska K, Śliwczyński A, Kotela I, Hordowicz M, Krakowiak J: Osteonecrosis related to steroid and alcohol use-an update on pathogenesis. Healthcare (Basel). 2023, 11:1846. 10.3390/healthcare11131846
- Chan KL, Mok CC: Glucocorticoid-induced avascular bone necrosis: diagnosis and management. Open Orthop J. 2012, 6:449-57. 10.2174/1874325001206010449
- Hartmann K, Koenen M, Schauer S, Wittig-Blaich S, Ahmad M, Baschant U, Tuckermann JP: Molecular actions of glucocorticoids in cartilage and bone during health, disease, and steroid therapy. Physiol Rev. 2016, 96:409-47. 10.1152/physrev.00011.2015
- Kaste SC, Karimova EJ, Neel MD: Osteonecrosis in children after therapy for malignancy. AJR Am J Roentgenol. 2011, 196:1011-8. 10.2214/AJR.10.6073
- Pierce TP, Jauregui JJ, Cherian JJ, Elmallah RK, Mont MA: Imaging evaluation of patients with osteonecrosis of the femoral head. Curr Rev Musculoskelet Med. 2015, 8:221-7. 10.1007/s12178-015-9279-6
- Gross CE, Haughom B, Chahal J, Holmes GB Jr: Treatments for avascular necrosis of the talus: a systematic review. Foot Ankle Spec. 2014, 7:387-97. 10.1177/1938640014521831
-
Thanks to AI, this year I was able to see a whole lot of zebras outside a hospital

-
1
-
-
Corcept Therapeutics Incorporated (NASDAQ: CORT), a commercial-stage company engaged in the discovery and development of medications to treat severe endocrinologic, oncologic, metabolic and neurologic disorders by modulating the effects of the hormone cortisol, today announced completion of enrollment in GRADIENT, a Phase 3 trial of its proprietary selective cortisol modulator relacorilant in patients with Cushing’s syndrome (hypercortisolism) caused by an adrenal adenoma or adrenal hyperplasia.
“Hypercortisolism with adrenal etiology affects many patients and is associated with serious cardiometabolic comorbidities, including hypertension and hyperglycemia, and increased risk of premature death,” said Bill Guyer, PharmD, Corcept’s Chief Development Officer. “GRADIENT is the first prospective placebo-controlled study to be conducted exclusively in these patients with Cushing’s syndrome. We expect data from GRADIENT in the fourth quarter of this year.”
GRADIENT is a randomized, double-blind, placebo-controlled trial conducted at sites in the United States, Europe and Israel. One-hundred thirty-seven patients were randomized 1:1 to receive relacorilant or placebo for 22 weeks. Primary endpoints are improvement in glucose metabolism and hypertension.
About Cushing’s Syndrome (Hypercortisolism)
Cushing’s syndrome is caused by excessive activity of the hormone cortisol. Endogenous Cushing’s syndrome is an orphan disease that most often affects adults aged 20-50. Symptoms vary, but most patients experience one or more of the following manifestations: high blood sugar, diabetes, high blood pressure, upper-body obesity, rounded face, increased fat around the neck, thinning arms and legs, severe fatigue and weak muscles. Irritability, anxiety, cognitive disturbances and depression are also common. Cushing’s syndrome can affect every organ system and can be lethal if not treated effectively.About Relacorilant
Relacorilant is a selective cortisol modulator that binds to the glucocorticoid receptor (GR), but does not bind to the body's other hormone receptors. Corcept is studying relacorilant in a variety of serious disorders, including ovarian, adrenal and prostate cancer and Cushing’s syndrome. Relacorilant is proprietary to Corcept and is protected by composition of matter, method of use and other patents. Relacorilant has orphan drug designation in the United States and the European Union for the treatment of Cushing’s syndrome.About Corcept Therapeutics
For over 25 years, Corcept’s focus on cortisol modulation and its potential to treat patients across a wide variety of serious disorders has led to the discovery of more than 1,000 proprietary selective cortisol modulators. Corcept’s advanced clinical trials are being conducted in patients with hypercortisolism, solid tumors, amyotrophic lateral sclerosis (ALS) and liver disease (NASH). In February 2012, the company introduced Korlym, the first medication approved by the U.S. Food and Drug Administration for the treatment of patients with Cushing’s syndrome. Corcept is headquartered in Menlo Park, California. For more information, visit Corcept.com.Forward-Looking Statements
Statements in this press release, other than statements of historical fact, are forward-looking statements based on our current plans and expectations that are subject to risks and uncertainties that might cause our actual results to differ materially from those such statements express or imply. These risks and uncertainties include, but are not limited to, our ability to operate our business; risks related to the study and development of Korlym as well as relacorilant, miricorilant, dazucorilant and our other product candidates, including their clinical attributes, regulatory approvals, mandates, oversight and other requirements; and the scope and protective power of our intellectual property. These and other risks are set forth in our SEC filings, which are available at our website and the SEC’s website.In this press release, forward-looking statements include those concerning the development of relacorilant as a treatment for Cushing’s syndrome, and design, timing and expectations regarding our GRADIENT trial. We disclaim any intention or duty to update forward-looking statements made in this press release.
From https://finance.yahoo.com/news/corcept-completes-enrollment-phase-3-120000179.html
-
1
-
-
Wow - a lot has happened since I first shared this post. If you look at the timestamp, I'm writing this at 4:20 am and I've been awake for an hour, even though I'm exhausted.
I have been back on Growth Hormone although it doesn't seem to do me any good.
I also had my knee replaced last March and I shared more about that in Bee’s Knees: TKR, Finally! I plan to get the other one done, presumably after next summer.

-
1
-
-
Comment from the blog:
Thank you Mary. I had surgery to remove pituitary tumor caused by Cushings in 2013. I am just starting to see some returning symptoms. Chronic stiff neck bloating belly gummy eyes. It feels like I am wearing a bras that is 5 times too small for me but I don’t have a bras on. A tightness across my back. 4 days ago one red spot showed up that I know is bleeding under the skin but I never knew the name of it. Now I know it is purpura because of this diagram. I know enough now that anything that looks strange on my body I take pictures of to show the doctor. Having your site is reassuring. I am still learning from it.
-
1
-
-
Even though this was from two years ago, I don't think that there have been any significant changes in the many symptoms of Cushing's.
Today's News Item proclaimed loftily
QuoteIn patients diagnosed with the disease yet who had inconclusive MRI results, PET/MRI was positive in 100% of cases, noted lead author Ilanah Pruis, a doctoral student at Erasmus University Medical Center in Rotterdam, Netherlands.
“This multimodal imaging technique provides a welcome improvement for diagnosis, planning of surgery, and clinical outcome in patients with Cushing disease,” the authors wrote.
But will enough doctors actually allow patients to get that far or will they still blow them off?
-
1
-
-
PET/MRI could become the diagnostic method of choice over MRI alone for identifying small pituitary tumors associated with Cushing disease, according to a study published March 21 in the Journal of Nuclear Medicine.
In patients diagnosed with the disease yet who had inconclusive MRI results, PET/MRI was positive in 100% of cases, noted lead author Ilanah Pruis, a doctoral student at Erasmus University Medical Center in Rotterdam, Netherlands.
“This multimodal imaging technique provides a welcome improvement for diagnosis, planning of surgery, and clinical outcome in patients with Cushing disease,” the authors wrote.
Cushing disease is characterized by small tumors in pituitary glands, which causes them to secrete excess cortisol, the authors explained. While it is a rare disease, over time it can cause severely disabling conditions, such as high blood pressure or type II diabetes.
Currently, guidelines recommend the use of MRI and inferior petrosal sinus sampling (IPSS) to diagnose these tumors. IPSS is an invasive procedure in which cortisol hormone levels are sampled from the veins that drain the pituitary gland.
In up to 40% of patients, however, MRI is inconclusive, as the lesions are smaller than 10 millimeters in diameter. Even advanced MRI techniques, such as dynamic perfusion imaging, can leave small lesions undetected in up to one third of patients, the authors noted.
In preclinical work, PET imaging using a radiotracer named F-18 FET has been shown to bind with high affinity to a molecular target in pituitary tumors, and in this study, the researchers aimed to test this technique combined with MRI in a multimodal approach.
The researchers analyzed results from 22 patients (68% women; mean age 48 years) who underwent F-18 FET PET/MRI at Erasmus MC between February 2021 and December 2022. All patients showed a clear pituitary tumor F-18 FET-PET/MRI, whereas reading of the MRI alone yielded a suspected lesion in only 50%, the authors found.
 T1-weighted postgadolinium MR images (A and C) and F-18 FET-PET/MR images (B and D) centered at pituitary before (A and
T1-weighted postgadolinium MR images (A and C) and F-18 FET-PET/MR images (B and D) centered at pituitary before (A and ") and after (C and D) transsphenoidal surgery. This patient with Cushing disease showed clear focal uptake (B) but no clear lesion on previously obtained and accompanying MRI (A). Postoperative tissue analysis did confirm resection of small pituitary adenoma/PitNET, and postoperative F-18 FET-PET showed no residual uptake (D). Image courtesy of the Journal of Nuclear Medicine.
and after (C and D) transsphenoidal surgery. This patient with Cushing disease showed clear focal uptake (B) but no clear lesion on previously obtained and accompanying MRI (A). Postoperative tissue analysis did confirm resection of small pituitary adenoma/PitNET, and postoperative F-18 FET-PET showed no residual uptake (D). Image courtesy of the Journal of Nuclear Medicine.
Importantly, 16 patients underwent treatment based on the results -- either surgery, Gamma Knife, or CyberKnife therapy -- with 12 of these patients achieving short-term remission, the authors noted.
“[F-18 FET-PET/MRI] is of great clinical value because it allows precision surgery and targeted Gamma Knife or CyberKnife therapy,” the group wrote.
The researchers noted that only one previous study evaluated F-18 FET-PET/MRI in these patients and that their study was limited, given the relatively small number of patients.
“Future studies will be directed at head-to-head comparisons of the performance of F-18 FET- PET and other diagnostic techniques, including advanced MRI sequences… preferably in patients at the time of initial clinical presentation,” the authors concluded.
A link to the full study can be found here.
-
 Please join in a Virtual Town Hall Meeting on Cushing's Awareness Day!
Please join in a Virtual Town Hall Meeting on Cushing's Awareness Day!
Mark your calendars for Monday, April 8, 2024, from 7 - 8 pm EST as we shed light on Cushing's syndrome with two incredible people who are living with this condition.
Gain valuable insights, hear personal stories, and learn more about Cushing's syndrome from those who understand it firsthand.
Don't miss this opportunity to connect, learn and show your support. Register now to secure your spot: https://www.eventbrite.com/.../cushings-awareness-day....
Let's come together to raise awareness and foster understanding. #CushingsAwareness
-
1
-

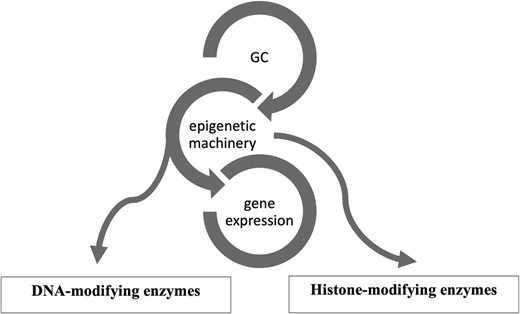
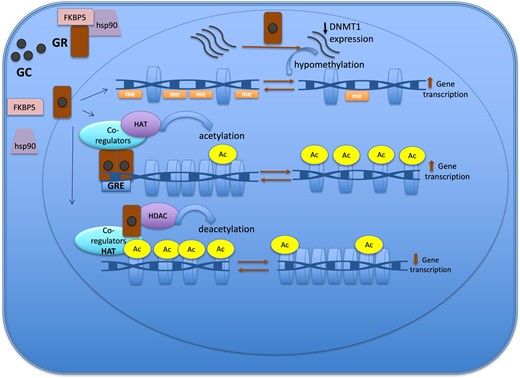



-shows-a-soft-tissue-lesion-located-in-the-midline-olfactory-groove-area.-Dural-surface-with-extension-into-anterior-frontal-dura.")
-shows-a-soft-tissue-lesion-located-in-the-midline-olfactory-groove-area.")
-demonstrates-three-focal-abnormal-uptakes:-the-largest-(5.2-x-2.4-cm)-in-the-left-submandibular-region,-and-two-smaller-ones-on-the-right,-suggestive-of-lymph-node-uptake.-Additional-abnormal-uptake-was-seen-along-the-midline-of-the-olfactory-groove-region-with-bilateral-extension.-No-other-significant-abnormal-uptake-was-identified.")


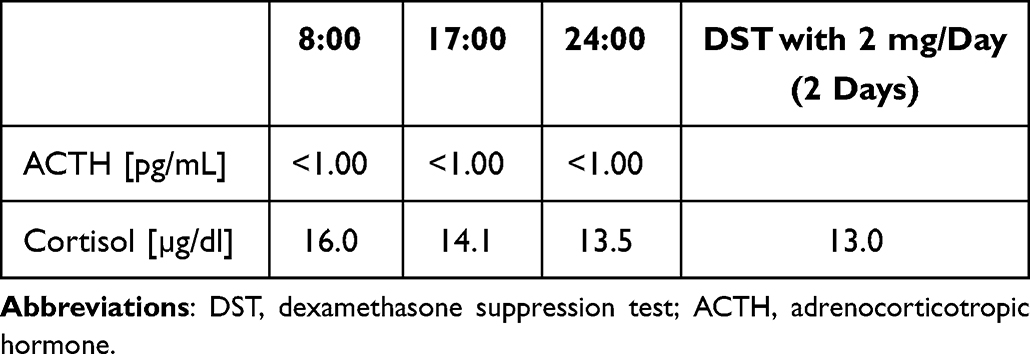
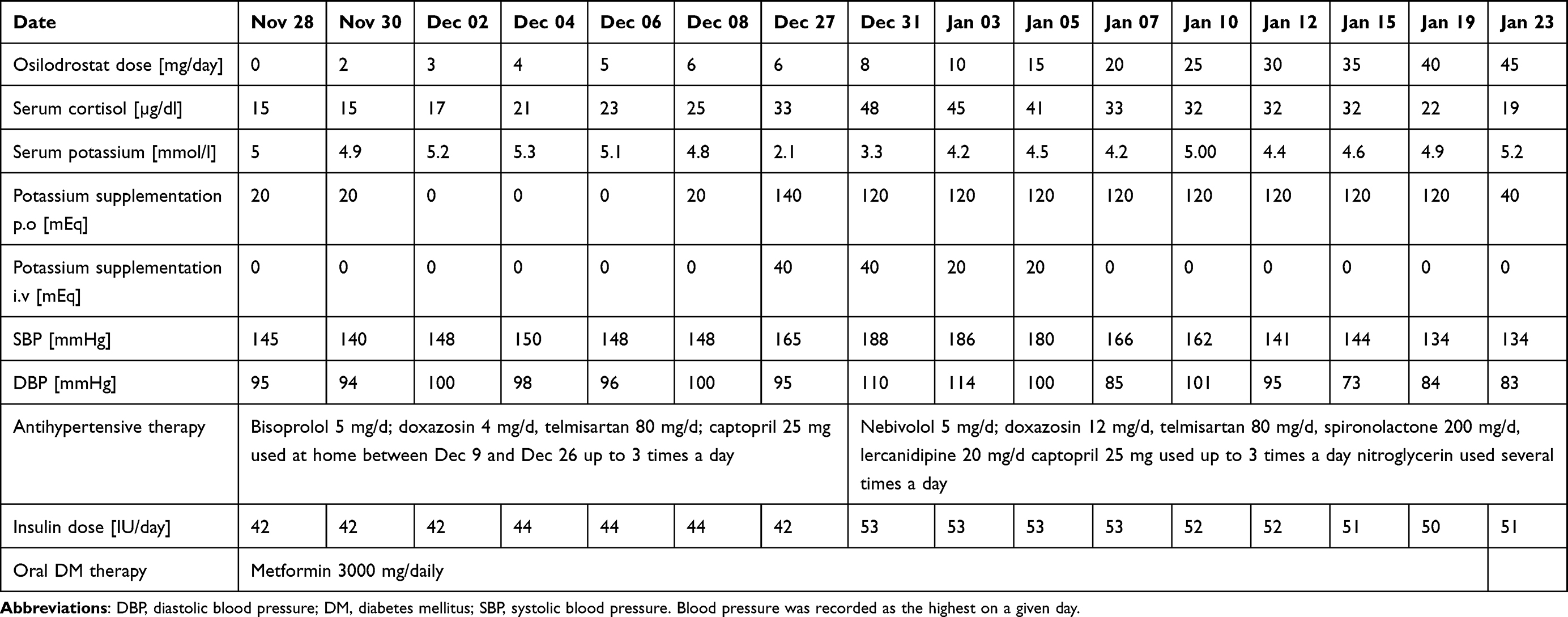
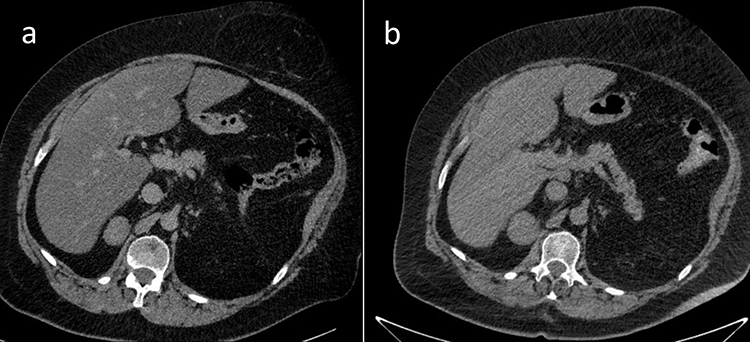
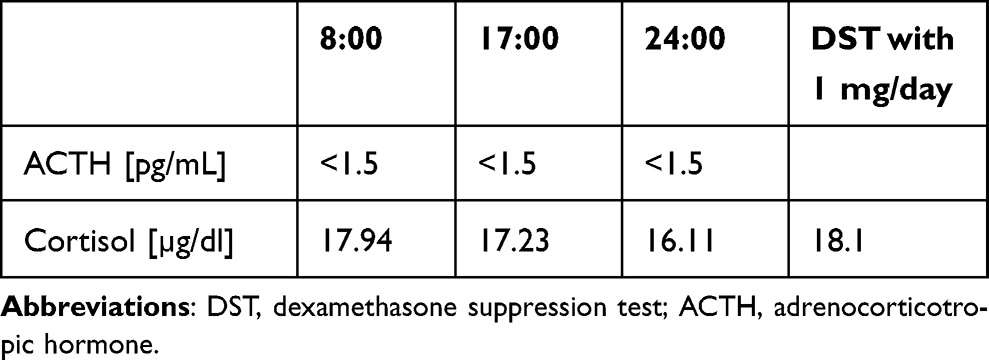
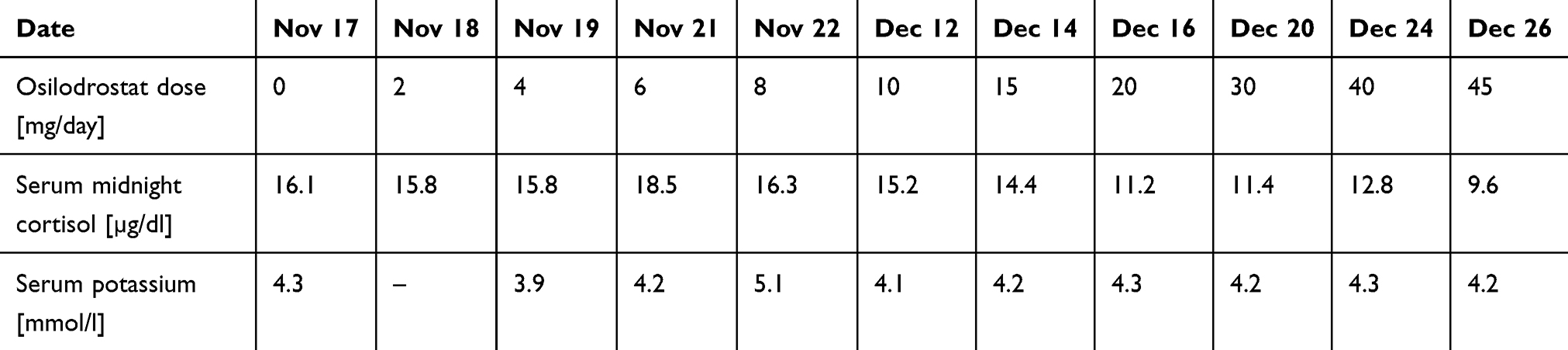



-bone-scan")





") and after (C and D) transsphenoidal surgery. This patient with Cushing disease showed clear focal uptake (B) but no clear lesion on previously obtained and accompanying MRI (A). Postoperative tissue analysis did confirm resection of small pituitary adenoma/PitNET, and postoperative F-18 FET-PET showed no residual uptake (D). Image courtesy of the
and after (C and D) transsphenoidal surgery. This patient with Cushing disease showed clear focal uptake (B) but no clear lesion on previously obtained and accompanying MRI (A). Postoperative tissue analysis did confirm resection of small pituitary adenoma/PitNET, and postoperative F-18 FET-PET showed no residual uptake (D). Image courtesy of the
{kind=link}
{kind=link}
{kind=link}
Day 17, Cushing’s Awareness Challenge
in Cushing's Awareness Day, April 8
Posted
I have seen this image several places online and it never ceases to crack me up. Sometimes, we really have strange things going on inside our bodies.
Usually, unlike Kermit, we ourselves know that something isn't quite right, even before the doctors know. Keep in touch with your own body so you'll know, even before the MRI.
I asked doctors for several years - PCP, gynecologist, neurologist, podiatrist - all said the now-famous refrain. "It's too rare. You couldn't have Cushing's." I kept persisting in my reading, making copies of library texts even when I didn't understand them, keeping notes. I just knew that someone, somewhere would "discover" that I had Cushing's.
Finally, someone did.
These days, there's no excuse to keep you from learning all you can about what's going on with you. There's your computer and the internet. Keep reading and learning all you can. You have a vested interest in what's going on inside, not your doctor.