

MaryO
-
Posts
8,066 -
Joined
-
Last visited
-
Days Won
549
Content Type
Profiles
Forums
Events
Blogs
Gallery
Articles
Media Demo
Store
Posts posted by MaryO
-
-
Introduction: The differential diagnosis between Cushing’s disease (CD) and ectopic ACTH syndrome (EAS) is complex, and bilateral inferior petrosal sinus sampling (BIPSS) is considered the gold-standard test. However, BIPSS with corticotropin-releasing hormone (CRH) stimulation is rarely available.
Objective: This retrospective cohort study aimed to assess the accuracy of the inferior petrosal sinus to peripheral ACTH gradient (IPS:P) before and after desmopressin stimulation for the differential diagnosis of ACTH-dependent Cushing’s syndrome (CS), applying different cutoff values.
Methods: A total of 50 patients (48 with CD and 2 with EAS) who underwent BIPSS were included in this study. The sensitivity and specificity of IPS:P in BIPSS before and after desmopressin stimulation were evaluated. Various cutoff values for IPS:P were examined to determine their diagnostic accuracy.
Results: Using the traditional IPS:P cutoff, the sensitivity was 85.1% before stimulation, 89.6% after stimulation, and a combined sensitivity of 91.7%. Applying cutoff values of IPS:P >1.4 before and >2.8 after stimulation, the sensitivity was 87.2% and 89.6%, respectively, with a combined sensitivity of 91.7%. Receiver operating characteristic (ROC) curve analysis determined optimal cutoff values of 1.2 before stimulation and 1.57 after stimulation, resulting in a sensitivity of 93.6% and 93.8%, respectively, with a combined sensitivity of 97.9%. Specificity remained at 100% throughout all analyses. Among the 43 patients who responded positively to stimulation, 42 (97.7%) did so within the first three minutes, and all 43 (100%) did so within the first five minutes. None of the assessed clinical variables predicted the ACTH response to stimulation in BIPSS with statistical significance.
Discussion: ACTH stimulation with desmopressin during BIPSS improves the accuracy of IPS:P, making it a valuable tool for investigating ACTH-dependent Cushing’s syndrome. Considering the low risk of complications, we recommend the use of desmopressin stimulation during BIPSS for the differential diagnosis of ACTH-dependent CS.
Introduction
Cushing Syndrome (CS) is a rare disease that results from chronic exposure to elevated cortisol levels. It can be caused by either endogenous or exogenous factors, and its incidence is estimated to be 0.7-3.2 cases per million per year (1, 2). The mortality rate for CS is elevated and may remain higher than the general population even after remission of hypercortisolism (3, 4). The causes of endogenous CS are traditionally classified into two categories: ACTH-dependent (about 80-85% of cases) and ACTH-independent (15-20% of cases) (5). The most common cause of ACTH-dependent CS (75-80% of cases) is Cushing Disease (CD), which is characterized by a corticotropic pituitary adenoma. The remaining cases (15-20%) of ACTH-dependent CS are caused by ectopic ACTH syndrome (EAS), which occurs when tumors of various sites, histological differentiation, and aggressiveness produce ACTH. There are also exceptionally rare cases (<1%) of ectopic CRH-producing tumors (5, 6).
CS diagnosis is a complex and challenging pathway due to the variable pattern of hormonal findings, the non-specificity of clinical presentation, particularly in mild hypercortisolism states (7), and the technical limitations of diagnostic tests. Once CS is confirmed, it should be differentiated between ACTH-dependent or -independent cases (8). ACTH levels <10 pg/ml suggest an adrenal cause; ACTH levels >20 pg/ml suggest ACTH-dependent causes; and levels between 10-20 pg/ml are considered indeterminate, requiring additional tests to establish the etiology (5, 8). When ACTH-dependency is confirmed, the next diagnostic step is the differentiation between CD and EAS. In this step, non-invasive tests are initially recommended, such as the CRH test (CRH-t), the 8 mg dexamethasone suppression test (DST-8 mg), and a pituitary magnetic resonance imaging (MRI) (5, 8). These tests, however, presents heterogenous results, depend on the availability of CRH, restricted in many countries including Brazil, and present low discriminatory power (9, 10). An alternative to CRH-t is the use of desmopressin, which stimulates ACTH release in most patients harboring ACTH-secreting pituitary adenomas. The use of this stimulus for the differential diagnosis of CD vs EAS is controversial, since studies have demonstrated that EAS patients may present ACTH elevation following desmopressin administration (11–14). The DST-8 mg is widely available; however it also presents limitation due to the variability of criteria used; furthermore, it has shown insufficient discriminatory capacity in some studies (15, 16). Pituitary MRI fails to detect adenomas in CD patients in about 30-50% of cases even with modern technology equipment (17); moreover, it may also generate false-positive results since pituitary incidentalomas are common in the population, including macroadenomas (18). In cases of conflicting non-invasive test results and unavailability of other methods, bilateral inferior petrosal sinus sampling (BIPSS) should be performed to detect a central-to-peripheral ACTH gradient that allows the localization of the ACTH production (5). Some authors and guidelines recommend performing BIPSS in all patients with pituitary lesions < 6 mm demonstrated on MRI (5, 8, 19), whereas others suggest BIPSS should routinely be performed, especially to guide surgical therapy of CD (20–23). Thus, the procedure is considered the gold-standard in the differential diagnosis of ACTH-dependent CS, preferentially performed with CRH or, less frequently, with desmopressin. The use of CRH is a limiting factor since it is unavailable in many countries. On the other hand, although used in some medical centers, desmopressin as a stimulus for BIPSS is still poorly debated and assessed in the literature, and its utility in this setting remains uncertain since studies validating it in different populations and in larger series are still lacking (8, 24–26). A recent study evaluating desmopressin in a large cohort of patients proposed new diagnostic criteria, questioning the need of stimulus with the new cut-offs (27). Thus, the aim of this study is to assess the role of central-to-peripheral ACTH gradient after stimulus with desmopressin during BIPSS for the differential diagnosis of ACTH-dependent CS in a cohort of patients followed-up in a referral center for CS in Brazil.
Patients and methods
Patients
Between 1998 and 2020, 107 patients with ACTH-dependent CS were retrospectively evaluated at the Neuroendocrinology clinic of a tertiary center in Southern Brazil for BIPSS under desmopressin stimulation during initial diagnostic evaluation or after recurrence. Of these, 58 patients underwent BIPSS with desmopressin, 50 of which for the initial diagnostic evaluation, 7 after recurrence and 1 after emergency adrenalectomy. Eight patients who underwent BIPSS were excluded for insufficient data regarding final etiologic diagnosis (lack of histopatological confirmation, lack of biochemical remission 6 months after surgery, or lack of remission after radiotherapy). Finally, 50 patients were included in the analysis. The present study was conducted in compliance with the principles laid down in the Declaration of Helsinki and was approved by the Hospital de Clínicas de Porto Alegre Ethics Committee.
Diagnosis of CS and ACTH-dependency status
After exhaustive screening for exogenous glucocorticoid administration, CS diagnosis was based on the presence of at least two of the following conditions: cortisol after low-dose dexamethasone suppression test (either 1 mg overnight or 0.5 mg 6/6 hours for 48h) > 1.8 µg/dL (DST-1mg); 24-h urinary free cortisol (UFC) or late night salivary cortisol consistently elevated in at least two samples (8). Additionally, late night serum cortisol > 7.5 µg/dL (8) and a desmopressin test (DES-t) with a peak ACTH > 71.8 pg/mL or an increase in ACTH ≥ 37 pg/mL from baseline (28) were also considered suggestive of CS.
After clinical and biochemical diagnostic confirmation of CS, plasma ACTH measurement classified CS into ACTH-dependent (ACTH > 20 pg/dL) or ACTH-independent (ACTH < 10 pg/dL). Values between 10-20 pg/dL were considered indeterminate and new samples were obtained for correct classification (8).
Next, patients diagnosed with ACTH-dependent CS underwent pituitary MRI for the identification of an adenoma. Due to the unavailability of CRH-t, it was rarely performed. The DES-t for the differential diagnosis of CD and EAS was considered predictive of CD when the increase was > 20% in cortisol or >35% in ACTH after stimulus. In virtue of its low accuracy, DST-8 mg was only performed in a few cases. Patients with inconclusive or negative imaging, those with adenomas < 6 mm or those with adenomas > 6 mm but discordant non-invasive tests were submitted to BIPSS with sampling of ACTH at baseline and after desmopressin stimulus.
After investigation, patients with a suggestive diagnosis of CD underwent transsphenoidal surgery. Histological confirmation of a pituitary adenoma staining positive for ACTH was considered the gold-standard for diagnosis. Additionally, patients with inconclusive or absent histological specimen who exhibited clinical and biochemical remission 6 months after surgery or who remitted after pituitary radiotherapy were also considered diagnosed for CD. The EAS cases were confirmed based on surgical excision or biopsy of tumoral lesions confirming the presence of ACTH-staining neoplastic cells.
Bilateral inferior petrosal sinus sampling
The procedure was performed in the presence of documented hypercortisolism, in an angiography room, under sedation with fentanyl and midazolam, and by a qualified professional in interventional radiology. Initially, bilateral common femoral venipuncture was performed, maintained with 6 French (F) introducers. Then, ascending catheterization of the superior vena cava and internal jugular veins was performed with a 5F vertebral catheter and hydrophilic guidewire, with final positioning of the catheter tip at the level of the inferior petrosal sinuses. Angiographic confirmation was performed after injection of 10 ml of diluted nonionic contrast under digital subtraction, demonstrating bilateral sinus and sellar region opacification. In situations of fine-caliber inferior petrosal sinuses, a coaxial microcatheter was used for a better distal reach of the required topography. Heparinization was not usually necessary in this technique, only sequential washing of the catheters was performed between the sampling times with saline solution with 2 ml of heparin for each 1000 ml of solution. Samples were collected after washing the catheters at baseline. Then, 10 µg of desmopressin was administered intravenously and samples were collected after one, three, five, and 15 minutes. In some cases, the sampling times were slightly different, but always with one sampling at baseline and at least 3 samplings after stimulation. All samples were collected in ice-cold tubes, kept on ice and then centrifuged in a refrigerated centrifuge and frozen at -8°C until ACTH measurement, which occurred immediately after the end of the procedure. After the samplings, the catheters and introducers were removed, followed by manual compression of the inguinal region at the puncture site for 10 minutes, until complete hemostasis. After compression, a compressive dressing was placed at the puncture site and the patients remained at bed rest without flexing the thigh for 6 h. Our routine protocol in performing the BIPSS did not include the concomitant measurement of prolactin as suggested in some previous studies in the literature.
Hormone assays
Until April 2004, cortisol was measured using a commercially available radioimmunoassay (RIA) kit (Diagnostic Systems Laboratories, Webster, TX, USA). From May 2004 to March 2010, the method was modified to an electrochemiluminescence immunoassay (ECLIA) kit (Modular Analytics E 170; Roche, Mannheim, Germany). From March 2010 to February 2014, cortisol was measured by chemiluminescence immunoassay (ADVIA Centaur XP Immunoassay System, Tarrytown, NY, USA). From February 2014 to October 2019, the method was Competitive Electrochemiluminescence. (Roche e602 equipment line). From October 2019 until the end of the study, the method was Microparticle Chemiluminescent Immunoassay. (Abbott equipment line). ACTH measurements up to February 2000 were performed by commercially available RIA. From February 2000 to April 2015, the method was chemiluminescence with the Immulite 1000 equipment. From May 2015 to April 2018, the method was electrochemiluminescence with the Roche e602 equipment. From May 2018 to August 2019, the method was sandwich electrochemiluminescence using the Roche e602 equipment. From August 2019 until the end of the study, the method was chemiluminescent immunoassay in the Immulite 2000 equipment. These assay differences do not show a large variation from normal values and as samples collected from the same patient were always analyzed with the same assay, the calculations of different indexes of central versus peripheral samplings did not change as a result of the trials. Of the cases studied, ACTH was measured by RIA in 1 patient, by Immulite 1000 in 35 patients, by Roche e602 via electrochemiluminescence in 9 patients, by Roche e602 via sandwich electrochemiluminescence in 4 patients and by Immulite 2000 in 1 patient.
The basal ACTH and UFC values, therefore, are presented according to the percentage above the ULN according to each methodology used at each moment. For the calculation of the ACTH inferior petrosal sinus to peripheral gradient (IPS:P), however, absolute values were used since the ratios are calculated for the same patient using the same assay.
Statistical analysis
The Kolmogorov-Smirnov test was used to assess the distribution of variables. Continuous variables with normal distribution are presented as mean ± standard deviation (SD). Continuous variables with asymmetric distribution are shown as median and interquartile range (IQR). Categorical variables were compared using Fischer’s exact test. The comparison of continuous variables was performed using the Mann-Whitney test. ROC curves were used to assess the ability of the IPS:P gradient to discriminate between CD and EAS, and the Youden index was used to define optimal cutoffs. Sensitivity and specificity were calculated for the different criteria analyzed. Statistical analyzes were performed using the SPSS 24.0 program (statistical package software, SPSS Incorporation, Chicago, IL, USA). Differences were considered significant when p<0.05.
Results
Patient characteristics are shown in Table 1. During the study period, 50 patients with a confirmed diagnosis of ACTH-dependent CS whose etiology could be confirmed through histopathological or biochemical data (remission after 6 months of surgery or after radiotherapy) who had undergone the BIPSS were included. The mean age (SD) at diagnosis was 38.22 (15.56) years, 39 patients (78%) were female, and 48 patients had CD and 2 EAS.
Table 1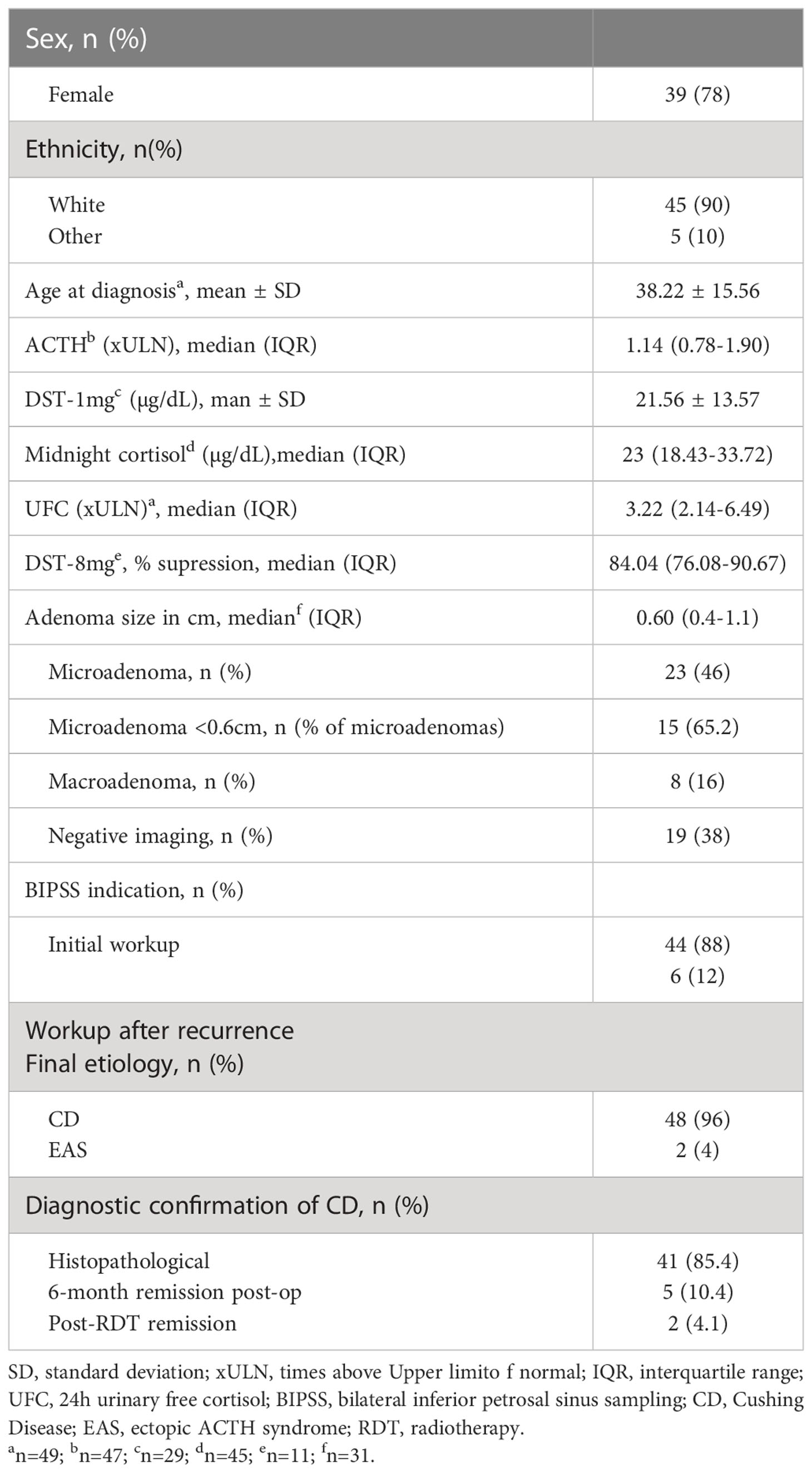
Table 1 Characteristics of studied patients.
In the imaging results, 23 (46%) were microadenomas, among which 15 were < 0.6cm (65.2% of microadenomas), 8 were macroadenomas (16%), and 19 had negative or inconclusive imaging (38%). One of the patients with EAS had an image suggestive of a 0.4 cm microadenoma on MRI. Regarding macroadenomas, the indication for BIPSS was proposed based on the following situations: 3 presented with a clinical picture of EAS, including 2 with systemic lesions suspicious for neoplasia, 3 presented imaging characteristics that were somewhat atypical for adenomas, 1 was associated with a brainstem vascular lesion and one was a recurrent disease with postsurgical alteration and residual lesion.
BIPSS was performed in 44 patients who had not yet undergone investigation or treatment and in 6 patients who had been previously treated for CD but had relapsed during follow-up. No complications were recorded in any of the cases submitted to BIPSS. There were no thromboembolism events related to the procedure.
At baseline (before stimulation), 49 patients were evaluated (1 patient with CD had samples collected, but his results were not properly recorded). The median IPS:P gradient at baseline was 6.62 (IQR 2.46-11.36) in patients with CD and 1.14 (IQR 1.10-1.14) in patients with EAS (p=0.01). Using the IPS:P>2 gradient criteria, 40 of 47 patients with CD were positive and none of the 2 patients with EAS were positive, resulting in 85.1% sensitivity (95% confidence interval (CI) 71.1-93.3%) and 100% specificity.
After stimulation with desmopressin, all 50 patients were evaluated. The median SPI:P gradient after stimulation was 29.46 (IQR 15.39-61.50) in patients with CD and 1.26 (min-max 1.25-1.28) in patients with EAS (p=0.01). In patients with EAS, the highest ACTH peak was 537 pg/mL (109.5% increase from baseline), while in patients with CD, the lowest increase from baseline was 19.48%. Using the IPS:P gradient criteria > 3, 43 of 48 patients with CD were positive, and none of the 2 patients with EAS were positive, resulting in 89.6% sensitivity (95%CI 76.5-96.1%) and 100% specificity. When evaluating patients who were positive at baseline and/or after stimulation in a combined manner, 44 of 48 CD patients were positive, whereas no EAS patients were positive. The overall sensitivity, therefore, was 91.7% (95%CI 79.1-97.3%), and the specificity was 100%. Of the 9 negative patients at baseline, 3 (33.33%) became positive after stimulation. Among the 43 patients who tested positive after the stimulus, 42 (97.7%) had already tested positive up to the third minute, and 100% of the patients were positive up to the fifth minute (Figure 1), totaling 86% of the total sample. Of the 3 patients whose stimulation was necessary, 2 had microadenomas and 1 had macroadenomas. In the two patients with EAS, the time of peak of ACTH was at 1 minute for patient 1 (31.1% increase from baseline) and at 3 minutes for patient 2 (109.5% increase from baseline).
Figure 1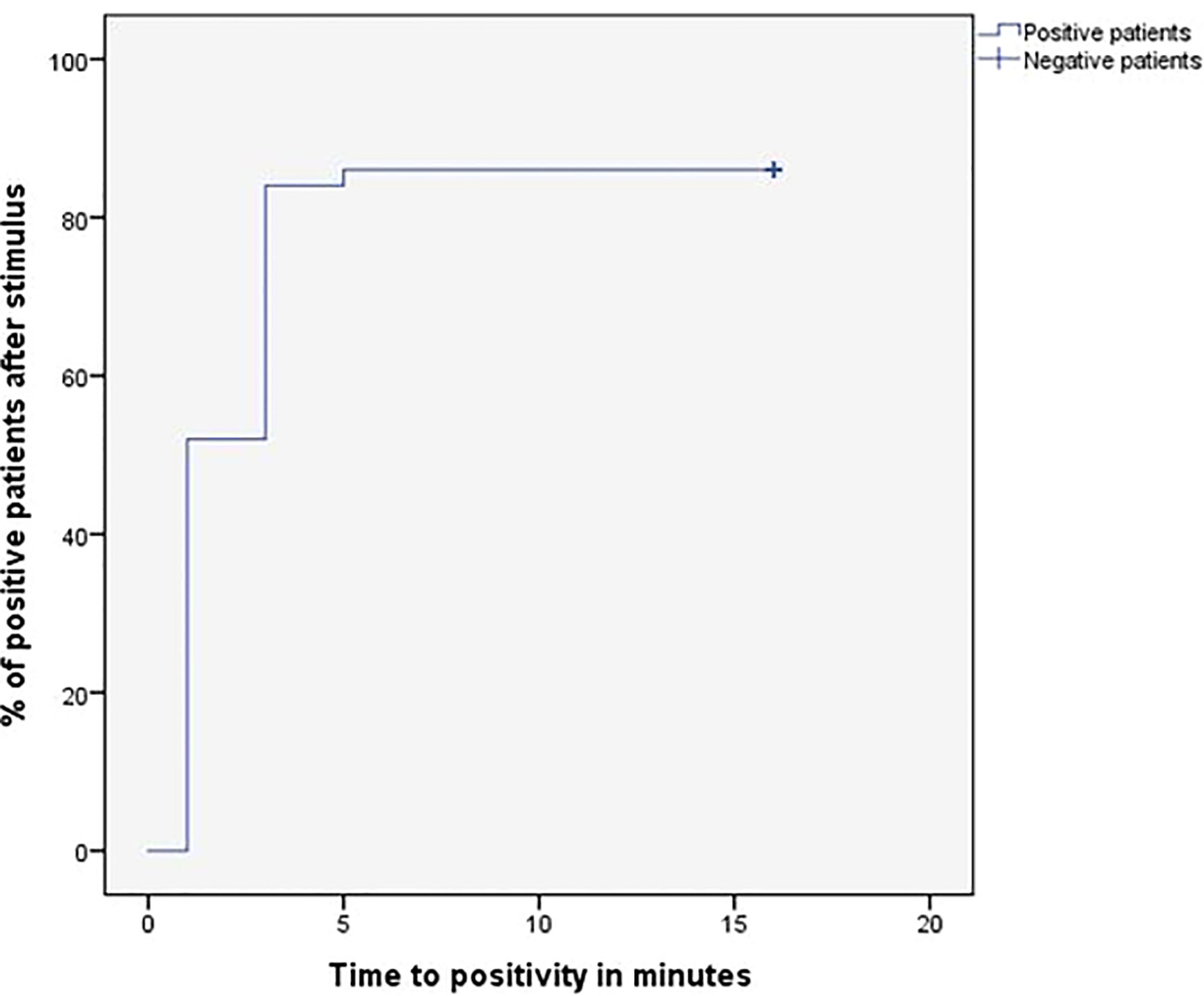
Figure 1 Time (minutes) until obtaining IPS:P gradient values of ACTH considered positive response of BIPSS after stimulation with desmopressin.
When assessing only the 23 patients with microadenoma, 20 of 22 patients with CD were positive at baseline, and the patient with EAS and 0.4 cm microadenoma was negative, resulting in 90.9% sensitivity (95%CI 69.37-98.4%), while maintaining 100% specificity. After stimulation, all 22 patients with CD were positive and the only patient with EAS and microadenoma was negative, resulting in 100% sensitivity (95%CI 81.5-100%) while maintaining 100% specificity. When only microadenomas < 0.6 cm were evaluated, 12 of 14 CD patients were positive at baseline, and the patient with EAS and 0.4 cm microadenoma was negative, resulting in 85.7% sensitivity (95%CI 56.2-97.5), with 100% specificity. After stimulation, all 14 patients with CD were positive, and the patient with EAS and microadenoma was negative, resulting in a sensitivity of 100% (95%CI 73.2-100%) while maintaining 100% specificity. All eight patients with microadenomas >0.6cm were already positive at baseline and remained positive after stimulation (100% sensitivity and 100% specificity). Thus, only patients with microadenoma <0.6 cm improved sensitivity after stimulation. Among the 8 patients with macroadenoma, sensitivity was 75% at baseline and remained the same after stimulation. However, when assessed for need for stimulation, only one patient with macroadenoma benefited, but sensitivity did not increase because a patient who was positive at baseline became negative after stimulation. Assessing all patients with positive imaging on MRI (micro or macroadenomas, n = 31), 26 of 30 CD patients were positive at baseline, and the patient with EAS and microadenoma was negative, resulting in 86.7% sensitivity and 100% specificity. After stimulation, 28 of 30 CD patients were positive and the patient with EAS and microadenoma remained negative, resulting in 93.3% sensitivity and maintaining 100% specificity. The combined sensitivity (baseline or after stimulus) in this group of patients was 96.7%.
Among the 19 patients with negative imaging, 18 had baseline results and were evaluated. Baseline sensitivity was 82.4%. After stimulation, data from 19 patients were evaluated and resulted in a sensitivity of 83.3%. When the patients with negative imaging (n=19) and those with microadenomas <0.6 cm (n=15) were analyzed together, which represent the most difficult cases in clinical practice, we observed that the IPS:P gradient >2 at baseline resulted in sensitivity of 83.9% and 100% specificity. After stimulation, the IPS:P >3 gradient had a sensitivity of 90.6% while maintaining 100% specificity.
After assessing the traditionally proposed criteria, the analysis was performed using the criteria proposed by Chen et al. (27). Using the IPS:P gradient at baseline > 1.4, 41 of 47 CD patients were positive and none of the EAS patients were positive, resulting in 87.2% sensitivity (95%CI 73.5-94.7%) while maintaining 100% specificity. After stimulation, using the IPS:P>2.8 gradient criteria, 43 of 48 patients with CD were positive, resulting in 89.6% sensitivity (95%CI 76.5-96.1%), strictly the same as the traditional criteria maintaining 100% specificity. When evaluating patients who were positive at baseline and/or after stimulation, 44 of 48 patients with CD were positive, and no patient with EAS was positive, resulting in 91.7% overall sensitivity (95%CI 79.1-97.3%), the same as the traditional criteria. Finally, only 2 of 49 patients who were negative at baseline became positive after stimulation.
To establish institution-specific cut-off points, a ROC curve was performed to assess the accuracy of the central/peripheral ACTH gradient in BIPSS in our cohort of patients. For the IPS:P gradient at baseline, the cut-off point with the highest accuracy was 1.2, whereas for the IPS:P gradient after stimulation, the cut-off point with the highest accuracy was 1.57 (Figure 2). Using these cut-off points, 44 of 47 CD patients were positive at baseline and no EAS patients were positive, resulting in 93.6% sensitivity (95%CI 81.4-98.3%), while maintaining 100% specificity. After stimulation, 45 of 48 CD patients were positive and no EAS patients were positive, resulting in 93.8% sensitivity (95%CI 81.8-98.4%), with 100% specificity (Figure 3). When evaluating patients who were positive at baseline and/or after stimulation, 47 of 48 CD patients were positive and no EAS patients were positive, resulting in an overall sensitivity of 97.9% (95%CI 87.5-99.9%) With 100% specificity. Finally, only 2 patients who were negative at baseline became positive after stimulation.
Figure 2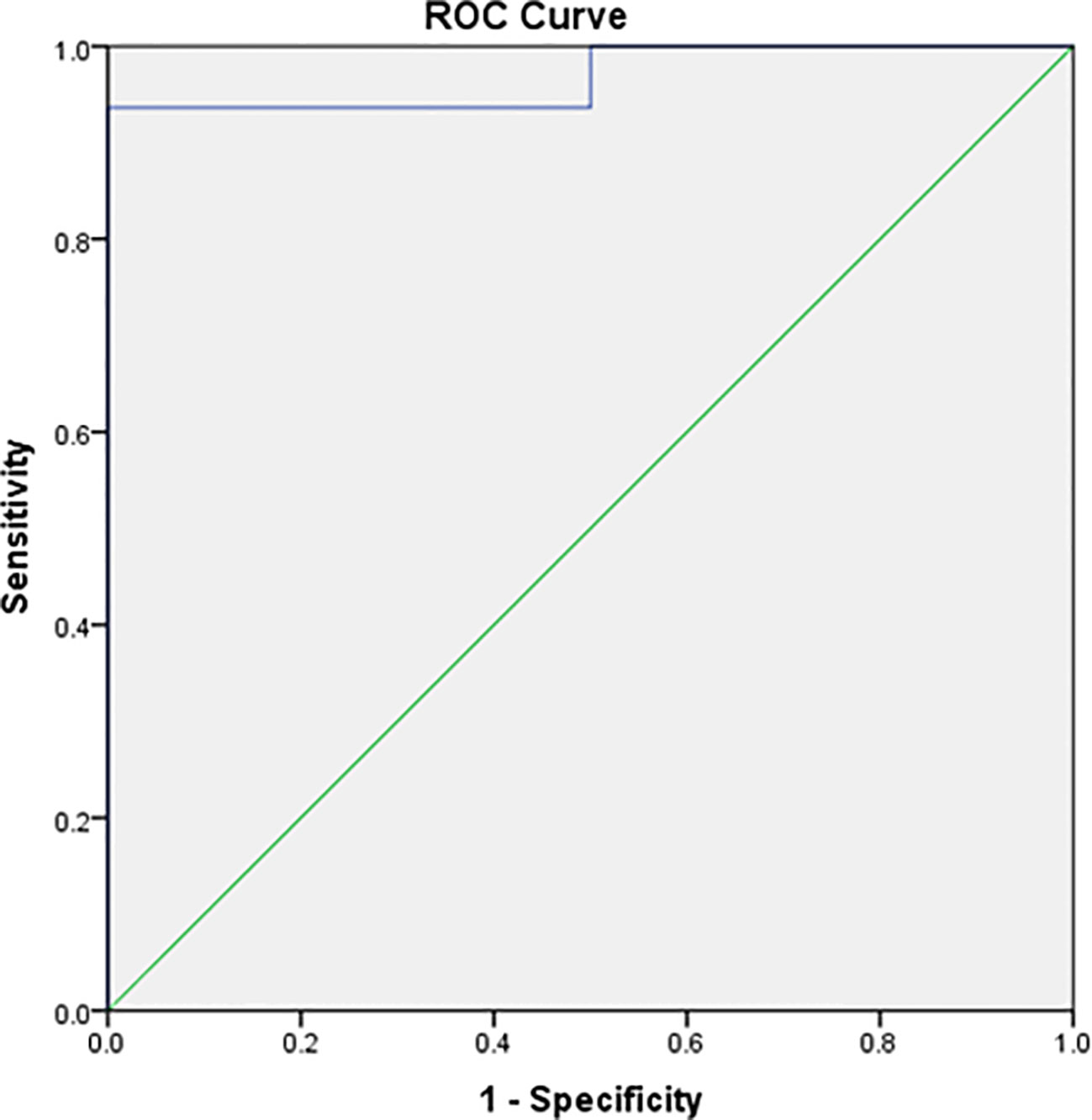
Figure 2 ROC curve of baseline IPS:P values in BIPSS in the investigation of ACTH-dependent CS.
Figure 3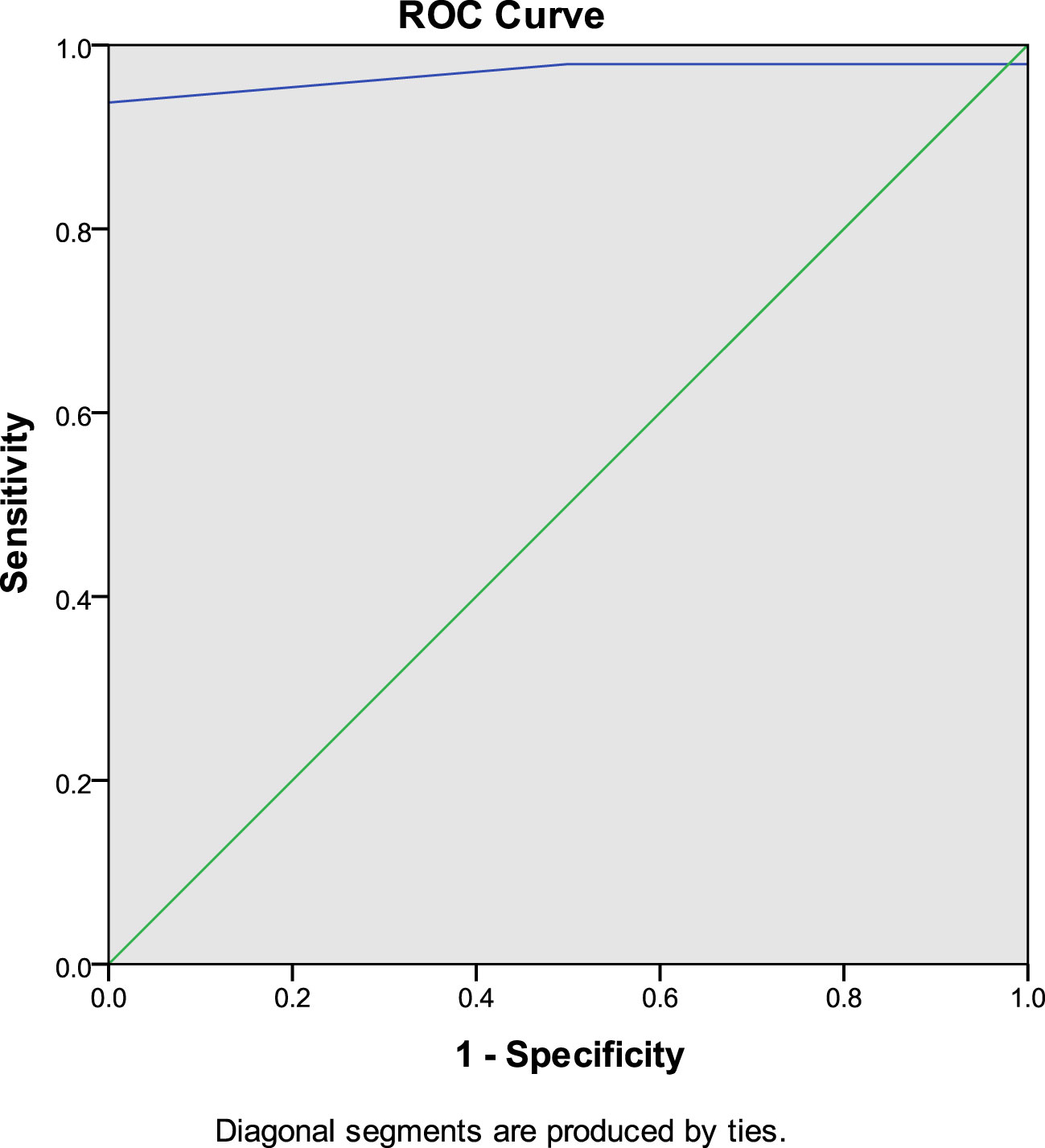
Figure 3 ROC curve of IPS:P values after stimulation with desmopressin in BIPSS in the investigation of ACTH-dependent CS.
In the comparison between the traditional criterion and our study criterion, the baseline sensitivity changed from 85.1 to 93.6%. After stimulation, baseline sensitivity changed from 89.6 to 93.8%, respectively. A summary of the sensitivity results with the different diagnostic criteria is presented in Table 2.
Table 2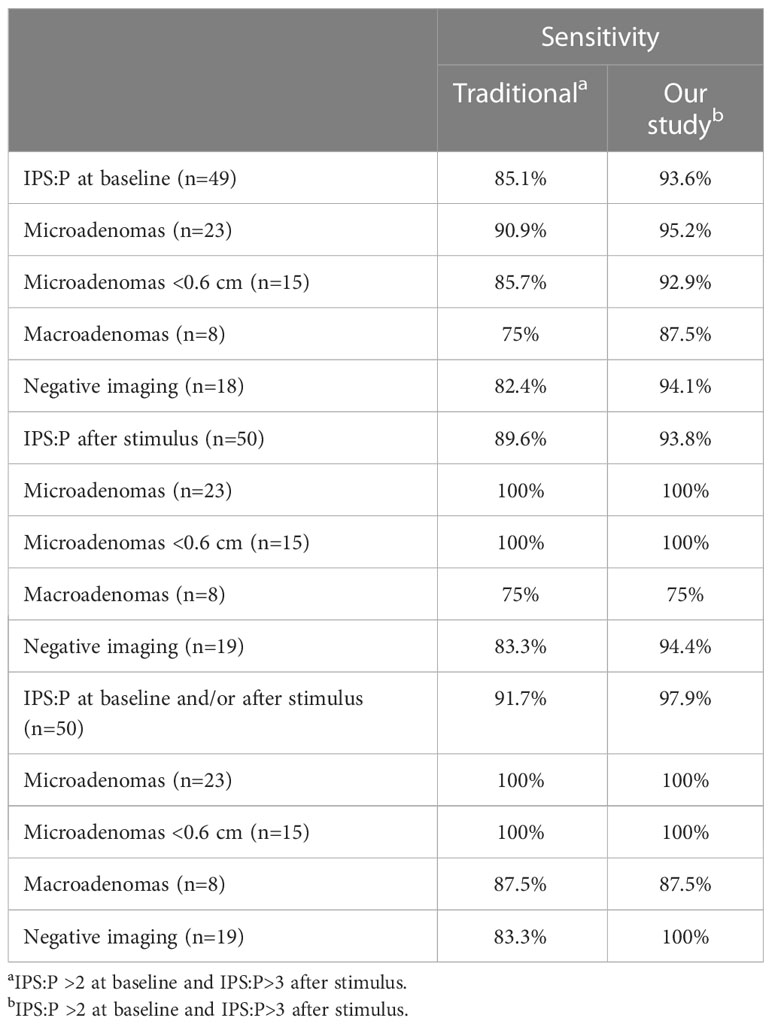
Table 2 Sensitivity of BIPSS with traditional criteria and with present study criteria.
Technical difficulties or anatomical variations were found in 6 patients undergoing BIPSS. Among the 43 cases with a positive IPS:P gradient, 3 had anatomical variations and 1 had some technical difficulty. Of the 5 cases in which the IPS:P gradient did not occur (false-negatives), 1 presented anatomical variation and 1 presented some technical difficulty during the test. Among the 6 patients who underwent BIPSS after recurrence, all had a final diagnosis of CD, and only 1 was negative on BIPSS.
Of the 50 patients evaluated, 43 had undergone DES-t as part of the diagnostic workup, of which 41 were later diagnosed with CD and 2 with EAS. Forty patients were considered responsive in DES-t, 38 patients with CD and 2 patients with EAS. Among the 40 responsive patients, 34 (85%) were also positive in BIPSS, all with a final diagnosis of CD. The 3 non-responsive patients in DES-t presented a positive response in BIPSS after desmopressin. Of the 6 patients who were positive in DES-t but negative in BIPSS, 2 were patients with EAS. Of the 4 patients with CD, 2 had normal petrosal sinus anatomy, 1 had a report of some anatomical variation, and 1 had a report of technical difficulties during BIPSS. Thus, DES-t was not able to predict response to desmopressin during BIPSS (p>0.9999). When comparing the ACTH values at baseline, 3, 5 and 10 minutes after stimulation in BIPSS, there was no significant difference between the group with positive versus negative DES-t, as well as no difference in the time to positivity between the groups, adenoma size, and number of patients with negative imaging. In addition, the clinical variables evaluated (ACTH, UFC, DST-1mg, baseline cortisol, adenoma size) were not able to significantly predict response to stimulus.
Discussion
In this study, the use of BIPSS with ACTH measurements at baseline and after stimulation with desmopressin in the differential diagnosis of the ACTH-producing source in a sample of 50 patients with ACTH-dependent CS and inconclusive non-invasive tests resulted in 85.1% baseline sensitivity, increasing to 89.6% after stimulation, maintaining 100% specificity when applying traditional IPS:P≥2 criteria at baseline and ≥3 after stimulation (29). When combined, the baseline and/or stimulated sensitivity results were 91.7%. Results of meta-analyses that combined studies performed with CRH stimulation and desmopressin indicate that the sensitivity of BIPSS ranges from 86-97% and the specificity from 89-100% (27, 30). Published studies with desmopressin are generally small, with a variable number of cases of EAS, different indications for BIPSS, and variable diagnostic criteria. In a study with a sample of 56 patients with ACTH-dependent CS and negative imaging, using the criterion of IPS:P≥2 at baseline and IPS:P≥3 after stimulation with desmopressin, the combined sensitivity was 92.1% and 100% specificity, similar to the findings of the present study (25). Smaller studies that also used desmopressin stimulation found similar (26, 31–33) or slightly higher sensitivities (34, 35). Studies performed exclusively in pediatric patients were less uniform, with one of them reporting similar results to studies that included adults (36) and another study demonstrating lower sensitivity in adult population (37).
Our institution’s optimal cut-off points, determined by analyzing the ROC curve, were IPS:P≥1.2 at baseline and ≥1.57 after stimulation. This resulted in 93.6% baseline sensitivity (it was 85.1% with IPS:P≥2), and 93.8% after stimulation (was 89.6% with IPS:P≥3), and a combined sensitivity of 97.9% (it was 91.7%), maintaining specificity at 100%. Despite the increased sensitivity, these criteria should be used with caution, since the number of cases with EAS was small. The IPS:P gradient at baseline and after stimulation achieved in patients with EAS in some studies with desmopressin would exceed the cutoffs found by us (24, 25, 27), which would incorrectly classify these patients as CD. Before adopting the new values in our institution, therefore, more patients with EAS are necessary to validate these criteria. Also using the ROC curve, Castinetti et al. evaluated 43 patients with ACTH-dependent SC (36 DC and 7 EAS) and established the criteria of IPS:P>2 at baseline or after stimulation, obtaining a sensitivity of 86% at baseline and 97% after stimulation with desmopressin, not mentioning the combined sensitivity. The study, however, showed 85% specificity at baseline, given that a patient with EAS had a 3.33 gradient (24). In addition to applying the traditional criteria, Machado et al. also used ROC curve analysis to establish cut-off points, finding an IPS:P≥1.45 at baseline (88.2% sensitivity) and ≥ 2.04 after stimulation (92.2% sensitivity) as optimal, both with 100% specificity, although the authors did not recommend the use of these new values (25). The results of these studies using the ROC curve suggest that lower cutoff points, both at baseline and after stimulation, can improve sensitivity without compromising specificity. However, a study that performed a ROC curve in patients stimulated with CRH found an optimal 2.10 baseline cut-off, slightly higher than the traditional one of 2, although the post-stimulation cut-off point was 2.15, lower than the one usually used (38). A study with desmopressin, in turn, found values in the ROC curve of 1.76 at baseline, lower than the traditional one, but ≥3.9 after stimulation, higher than the gradient of three usually used, increasing baseline sensitivity but keeping the sensitivity after stimulation unchanged (32).
The largest published study evaluating BIPSS with desmopressin stimulation evaluated 226 patients with CD and 24 with EAS (27). Applying the IPS:P>2 criteria at baseline and >3 after stimulation, the sensitivity was 87.2 and 94.2%, respectively, while maintaining 100% specificity. The combined sensitivity was 96.5%. In this series, 3 cases of EAS reached gradients greater than 2 after stimulation, which suggests that cut-off points equal to or lower than this may decrease specificity. The authors also performed an ROC curve, determining the cutoff point of >1.4 at baseline and >2.8 after stimulation. In this analysis, the sensitivity at baseline was 94.7% and 96% after stimulation, resulting in a combined 97.8% sensitivity, higher than that found with the traditional criteria. According to the authors, with these cut-off points, only 7 patients benefited from the stimulus. After this publication, no other studies have tested these new cutoffs. Our study was the first, therefore, to assess the new values. In our series, using the cutoff point of >1.4 at baseline and >2.8 after stimulation, the sensitivity was 87.2 and 89.3%, respectively, and the combined sensitivity was 91.7%, thus slightly improving the sensitivity at baseline with little change after stimulation.
In an attempt to identify predictors of need for stimulation, Chen et al. found that patients requiring stimulation had adenomas < 0.6 cm or negative imaging. In addition, patients who required stimulation had lower IPS ACTH levels and did not lateralize. These data, however, are obtained only after performing the BIPSS, which makes their use in practice unfeasible (27). In our series, among patients with microadenomas, only those with lesions <0.6 cm benefited from the stimulus. Patients with negative imaging had a small increase in sensitivity. A patient with a macroadenoma also benefited from the stimulus, although the sensitivity of the cases with macroadenoma did not change, as a positive patient at baseline became negative after the stimulus. Despite current recommendations suggesting to perform BIPSS in patients with adenomas < 0.6 cm or with negative/inconclusive imaging results (8, 39), Chen et al. identified 2 patients with EAS and adenomas > 0.6 cm who would be misdiagnosed with CD if the 0.6 cm threshold were respected. Therefore, they suggest performing BIPSS in all patients with ACTH-dependent CS (27). Given the relevance of EAS cases in this study, a discussion about the current size criteria for indicating BIPSS should be undertaken.
Of our 50 patients, 43 (41 CD and 2 EAS) underwent DES-t prior to BIPSS, and 40 were considered responsive, including the two cases of EAS. Among the responders, 34 patients also responded to the stimulus during the BIPSS, all of them with CD. The 3 patients who did not respond to the peripheral stimulus were, however, positive in the BIPSS. The lack of correlation between the DES-t results and the BIPSS may be related to the different sampling intervals in the two exams (short intervals in the BIPSS and long intervals in the peripheral test). Considering that the majority (86%) of our patients performed both tests, it is possible to conclude that the DES-t did not help in the prediction of response to the central stimulus, which makes the use of peripheral test results debatable for this purpose. Of the BIPSS studies with desmopressin, only one described the results of DES-t, although it did not perform any specific analysis of the relationship with BIPSS (36). The study differs from ours, also, as it only evaluated pediatric patients.
Although BIPSS is still considered the gold standard in the differential diagnosis of ACTH-dependent CS, some authors have suggested that the procedure should be indicated only in cases in which t-CRH was negative (40, 41). Recent studies have evaluated non-invasive strategies combining t-CRH, DES-t, TSD-8mg, and imaging to reduce the need for BIPSS. Strategies that resulted in a positive predictive value of 100%, however, included t-CRH as part of the diagnostic process (42, 43), which makes adherence to this diagnostic modality inapplicable in many countries due to the unavailability of CRH. In one of these studies, the combination of TSD-8mg with DES-t, which would be possible in Brazil, was inferior to the combination of DES-t with t-CRH or t-CRH with TSD-8mg (43). The low number of patients undergoing TSD-8mg in our study did not allow the evaluation of this strategy. Although not recommended as a test in the differential diagnosis of the etiology of ACTH-dependent SC, DES-t seems promising as a marker of long-term postoperative outcome and as an early marker of recurrence (44), which encourages further studies in these circumstances.
Despite there have been reports of thromboembolic events related to BIPSS that occurred heparin (45, 46), it is a very rare complication. The administration of desmopressin, which increases coagulation factor VIII and von Willebrand factor (47), has raised concerns about the potential for increased incidence of thromboembolic events during BIPSS. This is due to the fact that desmopressin is associated with the hypercoagulable state of CS (48) and may also interfere with VIII and von Willebrand factors. The study by Chen et al, the largest published with desmopressin to date, did not record any case of thromboembolism, even without routine anticoagulation during the procedure (27). In our study, performed without routine anticoagulation, there were also no thromboembolic events. The only desmopressin BIPSS study that recorded thromboembolic events routinely used heparin during the procedure (25). Thromboembolic events, therefore, do not appear to be an additional concern when using desmopressin, with or without the use of heparin during the procedure. The decision regarding the use or not of anticoagulants during BIPSS should be a decision of each institution and based on the usual anticoagulation recommendations.
In our study, we did not perform the concomitant dosage of prolactin in samples collected from the inferior petrosal sinuses, a procedure that potentially reduces false negatives, as advised by some authors based on studies with CRH (49–51) and a study with desmopressin (31). These findings, however, were not confirmed by all groups, both with CRH (52) and with desmopressin (32), and their applicability depends on further studies to define its role.
In our study, a total of 3 patients who were negative at baseline benefited from the stimulus, As they became positive, 2 of them with microadenomas and one with macroadenoma. The study by Chen et al. questions the use of routine stimulation in all patients to reduce the risks and the duration of the procedure, potentially reducing complications. The authors argue that, when using the IPS:P>1.4 criterion at baseline, the sensitivity was high enough to classify most patients, with the exception of 7 patients with adenoma <0.6 cm who needed stimulation (27). Our study, however, would have misclassified a case with CD and macroadenoma as EAS if the stimulus had not been performed. The assessment of the need for stimulation in cases of CS with macroadenoma is limited since most studies performed the BIPSS only in patients with lesions < 0.6 cm or negative imaging, preventing a more comprehensive assessment.
Considering that BIPSS is currently still the gold standard in the differential diagnosis of ACTH-dependent CS, even small gains in sensitivity should be considered important since incorrect classification of patients can lead to inappropriate treatments and potentially fatal delays in the resolution of hypercortisolism. Considering that BIPSS is generally well tolerated and the rate of serious complications is low (53), other strategies to reduce the risks of the procedure that do not involve avoiding the stimulus seem necessary. In this context, it is important to evaluate the time interval between the infusion of the secretagogue and the positive test result. In our study, 97.7% of the patients who tested positive after stimulation were already positive in the third minute and 100% of the patients were positive until the fifth minute, demonstrating that there seems to be no benefit in prolonging the test beyond this period. All of the few studies on BIPSS with desmopressin have directly or indirectly reported a similar time to positivity and for peak ACTH (i.e., positive up to 3-5 minutes) (26, 33, 35, 37). Stimulating patients for a maximum time of 5 minutes considerably reduces the procedure time without neglecting the sensitivity gain resulting from the stimulation and may, therefore, be a strategy to potentially reduce the risk of complications.
Our study evaluated a sample of patients whose BIPSS indication was more comprehensive since the unavailability of t-CRH and the low accuracy of TSD-8mg limited the use of non-invasive tests. The wide heterogeneity existing in the BIPSS studies regarding the characteristics of the evaluated patients (primary diagnosis or recurrence), the BIPSS technique (sampling times, anticoagulant use, material used, laboratory assays, cut-off points, type of secretagogue) makes direct comparisons difficult. Conducting multicenter prospective studies with a greater sample of EAS patients is necessary to improve our understanding of the best cut-off points and procedure duration.
The present study has some limitations, as expected in the complexity of CS investigation. Our main limitation is that the low prevalence of EAS that underwent BIPSS, resulting from the rarity of this condition, may explain the high specificity when applying the cutoff points indicated by the ROC curve, and the application of these new gradients of IPS:P depends on validation in larger samples of EAS. Lower specificity may result from poor responsiveness to the secretagogue (desmopressin or CRH), cyclic CS during periods of normal cortisol secretion or due to anomalous venous drainage (54). Retrospective data collection and analysis prevented access to complete information for all patients. There were differences over time in terms of sampling times, although at least 3 different samplings were always performed throughout the study period. We highlight that, in this study, we did not discuss the data regarding the eventual lateralization of the basal ACTH values and after stimulation with desmopressin to guide the location of the pituitary adenoma in the transsphenoidal surgery. This utility of the BIPSS has been less and less recommended in the literature due to the imprecision of the results, especially due to the existence of venous communications between the cavernous sinuses and the instability and intensity of blood aspiration for sample collection.
In conclusion, in BIPSS with ACTH dosage, the use of stimulation with desmopressin increases the sensitivity of the test from 85.1% to 89.6%, reaching 100% in the sub-analysis of microadenomas. In spite of being small, this increase is useful in the investigation of ACTH-dependent CS, a clinical situation in which gains in diagnostic sensitivity are very important. Additionally, considering the low risk of complications and the possibility to interrupt the test within 5 minutes, as demonstrated in our study, our data recommend the use of stimulation with desmopressin in the BIPSS in the differential diagnosis of ACTH-dependent CS.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving human participants were reviewed and approved by Hospital de Clínicas de Porto Alegre Ethics Committee. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
TA, TR and MC conceived the study and designed the research. TA conducted the data collection and database management. TA performed the data analysis. LS, MF and FG performed the BIPSS procedures. TA, TR, FC and MC contributed to the interpretation of the results. TA and MC drafted the manuscript. FC critically revised the manuscript. All authors read and approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Research Incentive Fund (FIPE) of Hospital de Clínicas de Porto Alegre and the Postgraduate Program in Medical Sciences: Endocrinology (PPG ENDO) from Universidade Federal do Rio Grande do Sul.
Acknowledgments
The authors would like to acknowledge the contributions of Guilherme Alcides Flores Soares Rollin, Arthur Boschi, and Camila Viecceli to the data collection process.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
BIPSS, bilateral inferior petrosal sinus sampling; CD, Cushing Disease; CRH-t, CRH test; CS, Cushing Syndrome; DES-t, desmopressin test; DST-1 mg, 1 mg dexamethasone suppression test; DST-8 mg, 8 mg dexamethasone suppression test; EAS, Ectopic ACTH Syndrome; ECLIA, electrochemiluminescence immunoassay; F, French; IPS:P, inferior petrosal sinus to peripheral gradient; IQR, interquartile range; MRI, magnetic resonance imaging; RIA, radioimmunoassay; SD, standard deviation; UFC, urinary free cortisol; ULN, upper limit of normal.
References
1. Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet (2006) 367(9522):1605–17. doi: 10.1016/S0140-6736(06)68699-6
2. Wengander S, Trimpou P, Papakokkinou E, Ragnarsson O. The incidence of endogenous Cushing’s syndrome in the modern era. Clin Endocrinol (Oxf) (2019) 91(2):263–70. doi: 10.1111/cen.14014
3. Ntali G, Asimakopoulou A, Siamatras T, Komninos J, Vassiliadi D, Tzanela M, et al. Mortality in Cushing’s syndrome: systematic analysis of a large series with prolonged follow-up. Eur J Endocrinol (2013) 169(5):715–23. doi: 10.1530/EJE-13-0569
4. Ragnarsson O, Olsson DS, Papakokkinou E, Chantzichristos D, Dahlqvist P, Segerstedt E, et al. Overall and disease-specific mortality in patients with cushing disease: A Swedish nationwide study. J Clin Endocrinol Metab (2019) 104(6):2375–84. doi: 10.1210/jc.2018-02524
5. Lacroix A, Feelders RA, Stratakis CA, Nieman LK. Cushing’s syndrome. Lancet (2015) 386(9996):913–27. doi: 10.1016/S0140-6736(14)61375-1
6. Sharma ST, Nieman LK, Feelders RA. Cushing’s syndrome: epidemiology and developments in disease management. Clin Epidemiol (2015) 7:281–93. doi: 10.2147/CLEP.S44336
7. Barbot M, Zilio M, Scaroni C. Cushing’s syndrome: Overview of clinical presentation, diagnostic tools and complications. Best Pract Res Clin Endocrinol Metab (2020) 34(2):101380. doi: 10.1016/j.beem.2020.101380
8. Machado MC, Fragoso MC, Moreira AC, Boguszewski CL, Vieira LN, Naves LA, et al. Recommendations of the Neuroendocrinology Department of the Brazilian Society of Endocrinology and Metabolism for the diagnosis of Cushing’s disease in Brazil. Arch Endocrinol Metab (2016) 60(3):267–86. doi: 10.1590/2359-3997000000174
9. Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab (2005) 90(8):4955–62. doi: 10.1210/jc.2004-2527
10. Isidori AM, Kaltsas GA, Grossman AB. Ectopic ACTH syndrome. Front Horm Res (2006) 35:143–56. doi: 10.1159/000094323
11. Newell-Price J, Perry L, Medbak S, Monson J, Savage M, Besser M, et al. A combined test using desmopressin and corticotropin-releasing hormone in the differential diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab (1997) 82(1):176–81. doi: 10.1210/jcem.82.1.3674
12. Terzolo M, Reimondo G, Ali A, Borretta G, Cesario F, Pia A, et al. The limited value of the desmopressin test in the diagnostic approach to Cushing’s syndrome. Clin Endocrinol (Oxf) (2001) 54(5):609–16. doi: 10.1046/j.1365-2265.2001.01260.x
13. Tsagarakis S, Tsigos C, Vasiliou V, Tsiotra P, Kaskarelis J, Sotiropoulou C, et al. The desmopressin and combined CRH-desmopressin tests in the differential diagnosis of ACTH-dependent Cushing’s syndrome: constraints imposed by the expression of V2 vasopressin receptors in tumors with ectopic ACTH secretion. J Clin Endocrinol Metab (2002) 87(4):1646–53. doi: 10.1210/jcem.87.4.8358
14. Suda T, Kageyama K, Nigawara T, Sakihara S. Evaluation of diagnostic tests for ACTH-dependent Cushing’s syndrome. Endocr J (2009) 56(3):469–76. doi: 10.1507/endocrj.K08E-353
15. Aron DC, Raff H, Findling JW. Effectiveness versus efficacy: the limited value in clinical practice of high dose dexamethasone suppression testing in the differential diagnosis of adrenocorticotropin-dependent Cushing’s syndrome. J Clin Endocrinol Metab (1997) 82(6):1780–5. doi: 10.1210/jc.82.6.1780
16. Isidori AM, Kaltsas GA, Mohammed S, Morris DG, Jenkins P, Chew SL, et al. Discriminatory value of the low-dose dexamethasone suppression test in establishing the diagnosis and differential diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab (2003) 88(11):5299–306. doi: 10.1210/jc.2003-030510
17. Vitale G, Tortora F, Baldelli R, Cocchiara F, Paragliola RM, Sbardella E, et al. Pituitary magnetic resonance imaging in Cushing’s disease. Endocrine (2017) 55(3):691–6. doi: 10.1007/s12020-016-1038-y
18. Freda PU, Beckers AM, Katznelson L, Molitch ME, Montori VM, Post KD, et al. Pituitary incidentaloma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab (2011) 96(4):894–904. doi: 10.1210/jc.2010-1048
19. Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2008) 93(5):1526–40. doi: 10.1210/jc.2008-0125
20. Findling JW, Kehoe ME, Shaker JL, Raff H. Routine inferior petrosal sinus sampling in the differential diagnosis of adrenocorticotropin (ACTH)-dependent Cushing’s syndrome: early recognition of the occult ectopic ACTH syndrome. J Clin Endocrinol Metab (1991) 73(2):408–13. doi: 10.1210/jcem-73-2-408
21. Graham KE, Samuels MH, Nesbit GM, Cook DM, O’Neill OR, Barnwell SL, et al. Cavernous sinus sampling is highly accurate in distinguishing Cushing’s disease from the ectopic adrenocorticotropin syndrome and in predicting intrapituitary tumor location. J Clin Endocrinol Metab (1999) 84(5):1602–10. doi: 10.1210/jcem.84.5.5654
22. Wiggam MI, Heaney AP, McIlrath EM, McCance DR, Sheridan B, Hadden DR, et al. Bilateral inferior petrosal sinus sampling in the differential diagnosis of adrenocorticotropin-dependent Cushing’s syndrome: a comparison with other diagnostic tests. J Clin Endocrinol Metab (2000) 85(4):1525–32. doi: 10.1210/jcem.85.4.6574
23. Kaskarelis IS, Tsatalou EG, Benakis SV, Malagari K, Komninos I, Vassiliadi D, et al. Bilateral inferior petrosal sinuses sampling in the routine investigation of Cushing’s syndrome: a comparison with MRI. AJR Am J Roentgenol (2006) 187(2):562–70. doi: 10.2214/AJR.05.0557
24. Castinetti F, Morange I, Dufour H, Jaquet P, Conte-Devolx B, Girard N, et al. Desmopressin test during petrosal sinus sampling: a valuable tool to discriminate pituitary or ectopic ACTH-dependent Cushing’s syndrome. Eur J Endocrinol (2007) 157(3):271–7. doi: 10.1530/EJE-07-0215
25. Machado MC, de Sa SV, Domenice S, Fragoso MC, Puglia P Jr., Pereira MA, et al. The role of desmopressin in bilateral and simultaneous inferior petrosal sinus sampling for differential diagnosis of ACTH-dependent Cushing’s syndrome. Clin Endocrinol (Oxf) (2007) 66(1):136–42. doi: 10.1111/j.1365-2265.2006.02700.x
26. Deipolyi AR, Alexander B, Rho J, Hirsch JA, Oklu R. Bilateral inferior petrosal sinus sampling using desmopressin or corticotropic-releasing hormone: a single-center experience. J Neurointerv Surg (2015) 7(9):690–3. doi: 10.1136/neurintsurg-2014-011262
27. Chen S, Chen K, Wang S, Zhu H, Lu L, Zhang X, et al. The optimal cut-off of BIPSS in differential diagnosis of ACTH-dependent cushing’s syndrome: is stimulation necessary? J Clin Endocrinol Metab (2020) 105(4):e1673–85. doi: 10.1210/clinem/dgz194
28. Rollin GA, Costenaro F, Gerchman F, Rodrigues TC, Czepielewski MA. Evaluation of the DDAVP test in the diagnosis of Cushing’s Disease. Clin Endocrinol (Oxf) (2015) 82(6):793–800. doi: 10.1111/cen.12661
29. Oldfield EH, Doppman JL, Nieman LK, Chrousos GP, Miller DL, Katz DA, et al. Petrosal sinus sampling with and without corticotropin-releasing hormone for the differential diagnosis of Cushing’s syndrome. N Engl J Med (1991) 325(13):897–905. doi: 10.1056/NEJM199109263251301
30. Wang H, Ba Y, Xing Q, Cai RC. Differential diagnostic value of bilateral inferior Petrosal sinus sampling (BIPSS) in ACTH-dependent Cushing syndrome: a systematic review and Meta-analysis. BMC Endocr Disord (2020) 20(1):143. doi: 10.1186/s12902-020-00623-3
31. Qiao X, Ye H, Zhang X, Zhao W, Zhang S, Lu B, et al. The value of prolactin in inferior petrosal sinus sampling with desmopressin stimulation in Cushing’s disease. Endocrine (2015) 48(2):644–52. doi: 10.1007/s12020-014-0338-3
32. Akbari H, Ghorbani M, Kabootari M, Mehrjardi AZ, Mohajeri Tehrani MR, Malek M, et al. Usefulness of prolactin measurement in inferior petrosal sinus sampling with desmopressin for Cushing’s syndrome. Br J Neurosurg (2020) 34(3):253–7. doi: 10.1080/02688697.2020.1736263
33. Salgado LR, Mendonça BB, Pereira MAA, Goic MSZ, Semer M, Moreira AC, et al. Use of desmopressin in bilateral and simultaneous inferior petrosal sinus sampling for differential diagnosis of ACTH-dependent cushing’s syndrome. Endocrinologist (1997) 7(3):135–40. doi: 10.1097/00019616-199707030-00001
34. Belli S, Oneto A, Mendaro E. [Bilateral inferior petrosal sinus sampling in the differential diagnosis of ACTH-dependent Cushing’s syndrome]. Rev Med Chil (2007) 135(9):1095–102. doi: 10.4067/s0034-98872007000900001
35. Feng M, Liu Z, Liu X, Zhang X, Bao X, Yao Y, et al. Tumour lateralization in Cushing’s disease by inferior petrosal sinus sampling with desmopressin. Clin Endocrinol (Oxf) (2018) 88(2):251–7. doi: 10.1111/cen.13505
36. Cavalcante LBCP, Freitas TC, Musolino NRC, Cescato VAS, Silva GO, Fragoso MCBV, et al. High accuracy of bilateral and simultaneous petrosal sinus sampling with desmopressin for the differential diagnosis of pediatric ACTH-dependent Cushing’s syndrome. Pituitary (2020) 23(5):507–14. doi: 10.1007/s11102-020-01051-1
37. Chen S, Chen K, Lu L, Zhang X, Tong A, Pan H, et al. The effects of sampling lateralization on bilateral inferior petrosal sinus sampling and desmopressin stimulation test for pediatric Cushing’s disease. Endocrine (2019) 63(3):582–91. doi: 10.1007/s12020-018-1779-x
38. Colao A, Faggiano A, Pivonello R, Pecori Giraldi F, Cavagnini F, Lombardi G, et al. Inferior petrosal sinus sampling in the differential diagnosis of Cushing’s syndrome: results of an Italian multicenter study. Eur J Endocrinol (2001) 144(5):499–507. doi: 10.1530/eje.0.1440499
39. Nieman LK, Biller BM, Findling JW, Murad MH, Newell-Price J, Savage MO, et al. Treatment of cushing’s syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab (2015) 100(8):2807–31. doi: 10.1210/jc.2015-1818
40. Zampetti B, Grossrubatscher E, Dalino Ciaramella P, Boccardi E, Loli P. Bilateral inferior petrosal sinus sampling. Endocr Connect (2016) 5(4):R12–25. doi: 10.1530/EC-16-0029
41. Losa M, Allora A, Panni P, Righi C, Mortini P. Bilateral inferior petrosal sinus sampling in adrenocorticotropin-dependent hypercortisolism: always, never, or sometimes? J Endocrinol Invest (2019) 42(8):997–1000. doi: 10.1007/s40618-019-1006-5
42. Frete C, Corcuff JB, Kuhn E, Salenave S, Gaye D, Young J, et al. Non-invasive diagnostic strategy in ACTH-dependent cushing’s syndrome. J Clin Endocrinol Metab (2020) 105(10):3273–84. doi: 10.1210/clinem/dgaa409
43. Ferrante E, Barbot M, Serban AL, Ceccato F, Carosi G, Lizzul L, et al. Indication to dynamic and invasive testing in Cushing’s disease according to different neuroradiological findings. J Endocrinol Invest (2022) 45(3):629–37. doi: 10.1007/s40618-021-01695-1
44. Vassiliadi DA, Tsagarakis S. DIAGNOSIS OF ENDOCRINE DISEASE: The role of the desmopressin test in the diagnosis and follow-up of Cushing’s syndrome. Eur J Endocrinol (2018) 178(5):R201–R14. doi: 10.1530/EJE-18-0007
45. Obuobie K, Davies JS, Ogunko A, Scanlon MF. Venous thrombo-embolism following inferior petrosal sinus sampling in Cushing’s disease. J Endocrinol Invest (2000) 23(8):542–4. doi: 10.1007/BF03343772
46. Blevins LS Jr., Clark RV, Owens DS. Thromboembolic complications after inferior petrosal sinus sampling in patients with cushing’s syndrome. Endocr Pract (1998) 4(6):365–7. doi: 10.4158/EP.4.6.365
47. Kaufmann JE, Vischer UM. Cellular mechanisms of the hemostatic effects of desmopressin (DDAVP). J Thromb Haemost (2003) 1(4):682–9. doi: 10.1046/j.1538-7836.2003.00190.x
48. van der Pas R, Leebeek FW, Hofland LJ, de Herder WW, Feelders RA. Hypercoagulability in Cushing’s syndrome: prevalence, pathogenesis and treatment. Clin Endocrinol (Oxf) (2013) 78(4):481–8. doi: 10.1111/cen.12094
49. Findling JW, Kehoe ME, Raff H. Identification of patients with Cushing’s disease with negative pituitary adrenocorticotropin gradients during inferior petrosal sinus sampling: prolactin as an index of pituitary venous effluent. J Clin Endocrinol Metab (2004) 89(12):6005–9. doi: 10.1210/jc.2004-1378
50. Mulligan GB, Eray E, Faiman C, Gupta M, Pineyro MM, Makdissi A, et al. Reduction of false-negative results in inferior petrosal sinus sampling with simultaneous prolactin and corticotropin measurement. Endocr Pract (2011) 17(1):33–40. doi: 10.4158/EP10067.OR
51. Grant P, Dworakowska D, Carroll P. Maximizing the accuracy of Inferior petrosal sinus sampling: validation of the use of Prolactin as a marker of pituitary venous effluent in the diagnosis of Cushing’s disease. Clin Endocrinol (Oxf) (2012) 76(4):555–9. doi: 10.1111/j.1365-2265.2011.04257.x
52. De Sousa SMC, McCormack AI, McGrath S, Torpy DJ. Prolactin correction for adequacy of petrosal sinus cannulation may diminish diagnostic accuracy in Cushing’s disease. Clin Endocrinol (Oxf) (2017) 87(5):515–22. doi: 10.1111/cen.13401
53. Vassiliadi DA, Mourelatos P, Kratimenos T, Tsagarakis S. Inferior petrosal sinus sampling in Cushing’s syndrome: usefulness and pitfalls. Endocrine (2021) 73(3):530–9. doi: 10.1007/s12020-021-02764-4
Keywords: Cushing’s syndrome, Cushing’s disease, ectopic ACTH syndrome, bilateral inferior petrosal sinus sampling, ACTH, desmopressin
Citation: Almeida TSd, Rodrigues TdC, Costenaro F, Scaffaro LA, Farenzena M, Gastaldo F and Czepielewski MA (2023) Enhancing Cushing’s disease diagnosis: exploring the impact of desmopressin on ACTH gradient during BIPSS. Front. Endocrinol. 14:1224001. doi: 10.3389/fendo.2023.1224001
Received: 17 May 2023; Accepted: 11 July 2023;
Published: 03 August 2023.Edited by:
Fabienne Langlois, Centre Hospitalier Universitaire de Sherbrooke, CanadaReviewed by:
Filippo Ceccato, University of Padua, Italy
Matthieu St-Jean, Université de Sherbrooke, CanadaCopyright © 2023 Almeida, Rodrigues, Costenaro, Scaffaro, Farenzena, Gastaldo and Czepielewski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tobias Skrebsky de Almeida, tsalmeid@gmail.com
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
From https://www.frontiersin.org/articles/10.3389/fendo.2023.1224001/full
-
 1
1
-
-
Background: Café-au-lait skin macules, Cushing syndrome (CS), hyperthyroidism, and liver and cardiac dysfunction are presenting features of neonatal McCune–Albright syndrome (MAS), CS being the rarest endocrine feature. Although spontaneous resolution of hypercortisolism has been reported, outcome is usually unfavorable. While a unified approach to diagnosis, treatment, and follow-up is lacking, herein successful treatment and long-term follow-up of a rare case is presented.
Clinical case: An 11-day-old girl born small for gestational age presented with deterioration of well-being and weight loss. Large hyperpigmented macules on the trunk, hypertension, hyponatremia, hyperglycemia, and elevated liver enzymes were noted. ACTH-independent CS due to MAS was diagnosed. Although metyrapone (300 mg/m2/day) was started on the 25th day, complete remission could not be achieved despite increasing the dose up to 1,850 mg/m2/day. At 9 months, right total and left three-quarters adrenalectomy was performed. Cortisol decreased substantially, ACTH remained suppressed, rapid tapering of hydrocortisone to physiological dose was not tolerated, and supraphysiological doses were required for 2 months. GNAS analysis from the adrenal tissue showed a pathogenic heterozygous mutation. During 34 months of follow-up, in addition to CS due to MAS, fibrous dysplasia, hypophosphatemic rickets, and peripheral precocious puberty were detected. She is still regularly screened for other endocrinopathies.
Conclusion: Neonatal CS due to MAS is extremely rare. Although there is no specific guideline for diagnosis, treatment, or follow-up, addressing side effects and identifying treatment outcomes will improve quality of life and survival.
Introduction
McCune–Albright syndrome (MAS) is a rare mosaic disorder of remarkable complexity with an estimated prevalence of 1/100,000 and 1/1,000,000 (1). Timing of postzygotic missense gain of function mutation of GNAS encoding stimulatory Gαs determines the extent of tissue involvement, imposing a unique clinical phenotype. Although a combination of two or more classical features, such as fibrous dysplasia of bone (FD), café-au-lait skin macules, and hyperfunctioning endocrinopathies (gonadotropin-independent gonadal function, nonautoimmune hyperthyroidism, growth hormone excess, and neonatal hypercortisolism), are diagnostic, renal, hepatobiliary, and cardiac involvement have also been reported (2–4).
Adrenocorticotropic hormone (ACTH)-independent adrenal Gαs activation results in the rarest endocrine feature of MAS, which almost invariably presents in the neonatal period: Cushing syndrome (CS). Due to greater burden of Gαs-mutation-bearing cells, the presence of CS is correlated with increased number of accompanying features of MAS and a poorer outcome. Although there is spontaneous resolution in 33% of cases with neonatal CS, mortality occurs with a high rate of 20% (4).
A dilemma for the clinician is that most publications to date have been case reports, and there is as yet no guideline for diagnosis, treatment, or follow-up. Here, a rare case of severe CS due to MAS, underlining the unique clinical phenotype specific to the neonatal period, is presented. Our goal is to offer a practical approach based on 3 years of clinical experience of this rare disorder that will help navigate challenges during follow-up.
Case presentation
A baby girl, born small for gestational age with a birthweight of 2,340 g (−2.1 SDS) and a head circumference of 32.6 cm (−1.61 SDS) was admitted to the neonatal intensive care unit in the first day of life for respiratory distress. She was the second child of a healthy non-consanguineous Caucasian couple, born 38 weeks of gestation via cesarean section following an uneventful pregnancy. Alanine aminotransferase [ALT, 2,376 U/L (normal, 0–40)] and aspartate aminotransferase [AST, 875 U/L (normal, 0–40)] were elevated; gamma-glutamyl transferase and bilirubin were normal. Antibiotics were administered intravenously after a diagnosis of possible neonatal sepsis. Respiratory distress resolved, and liver enzymes decreased (ALT, 687 U/L; AST, 108 U/L). As soon as the antimicrobial treatment was completed, she was discharged in the seventh day of life.
She was referred to our center, 4 days later, for failure to thrive (2,315 g), difficulty in feeding, and deterioration of general health. On physical examination, round facies, elongated philtrum and retro-micrognatia, hyperpigmented macules both at the front and back of the trunk and on labia majora, which do not cross midline, and hypertrichosis on the forehead and extremities were noted (Supplementary Figure S1). Newborn reflexes were hypoactive, blood pressure was 100/70 mmHg, and second-degree cardiac murmur was also detected. Systems were normal otherwise. Laboratory findings revealed hyponatremia, impaired renal and liver function tests, tubulopathy, and proteinuria, while blood count was normal (hemoglobin, 10.4 g/dl; leukocyte, 25.0 × 103/μl; platelet count, 449×103/μl) (Table 1). Hyponatremia resolved with fluid treatment, while liver enzymes, blood urea nitrogen, and creatinine remained elevated. Further endocrine evaluation revealed an elevated serum basal cortisol [225.68 g/dl (N, 6.7–22.6 µg/dL)] and 24-h urinary free cortisol [1,129 μg/day (N, 1.4–20 μg/day)]. Serum cortisol was not suppressed during overnight high-dose dexamethasone suppression test (Table 2) (5). Thyroid hormones were consistent with non-thyroidal illness.
Table 1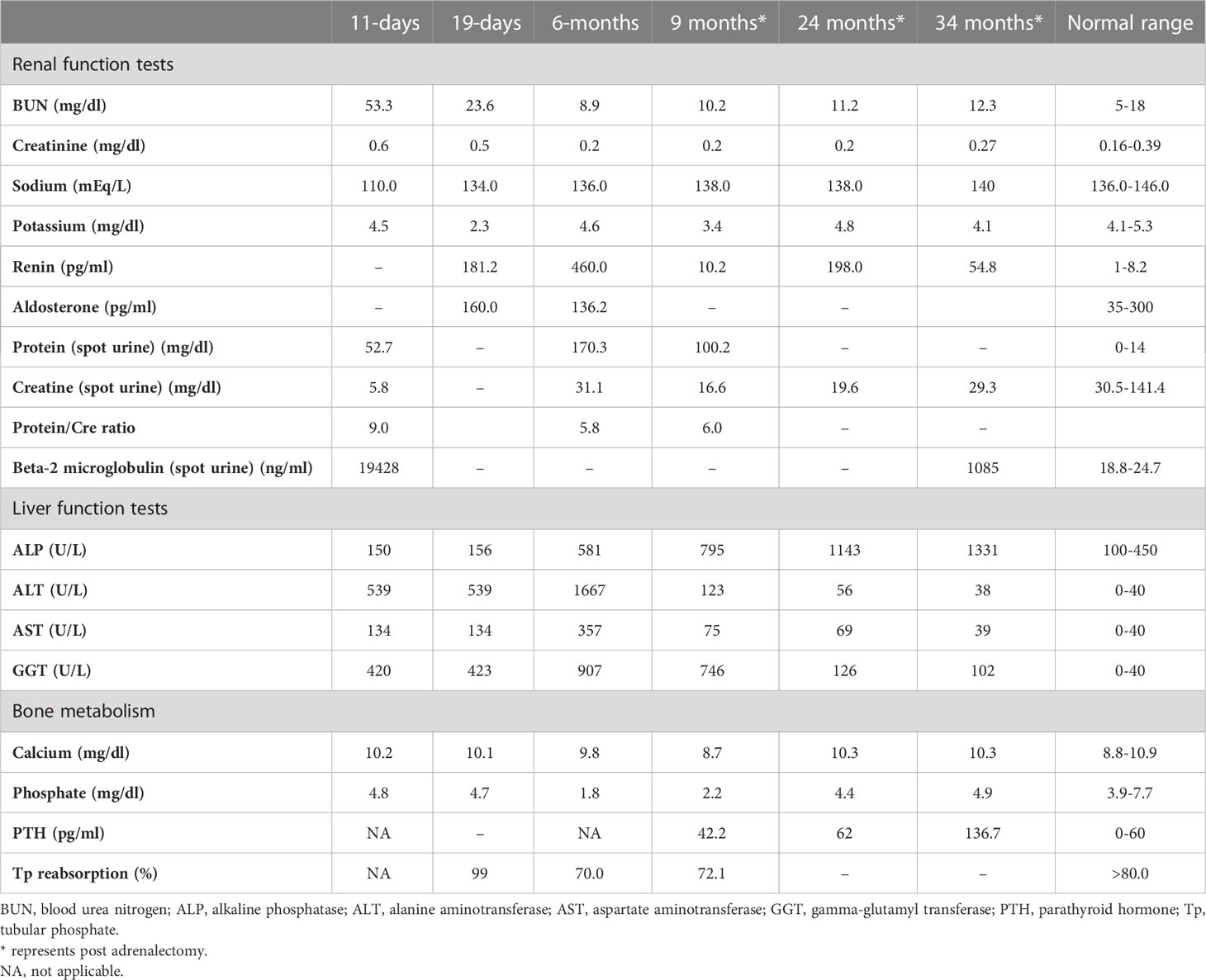
Table 1 Laboratory investigations on admission, prior to medical treatment (19 days), after medical treatment (6 months), and post-adrenalectomy.
Table 2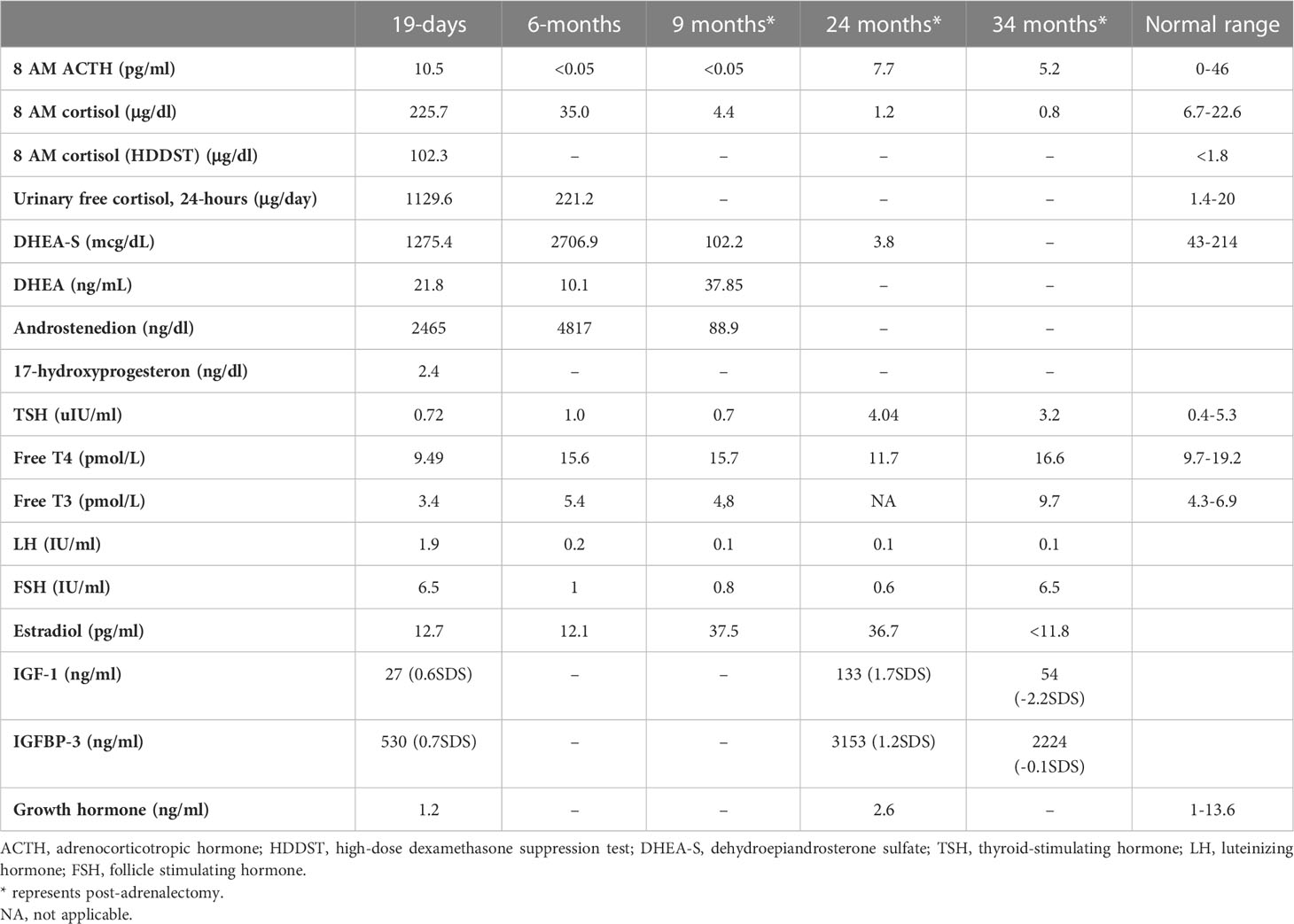
Table 2 Endocrine evaluation prior to medical treatment (19 days), after medical treatment (6 months), and post-adrenalectomy.
ACTH-independent CS and café-au-lait spots suggested MAS. Hypercortisolism-related complications emerged. On the 11th day, hyperglycemia (blood glucose, 250 mg/dl) was seen, and it persisted after cessation of intravenous fluids in the exclusively breastfed neonate; thus, 0.5 U subcutaneous neutral protamine Hagedorn insulin (NPH) (three times a day) was initiated on the 16th day of life when blood glucose was 340 mg/dl, and serum insulin was 18.10 μIU/ml. Hypertension (110/90 mmHg) and hypokalemia were triggered by mineralocorticoid action of excessive cortisol on 20th day. Spironolactone (2 mg/kg/day) was started, and nifedipine (0.5 mg/kg/day) was added in order to control blood pressure (Supplementary Figure S2). Since immunosuppressive effects of excess cortisol may increase the risk for opportunistic infections, Pneumocystis jirovecii prophylaxis was started and live vaccines were postponed.
Features of MAS and accompanying hyperfunctioning endocrinopathies were screened (Table 2). On ultrasonography, adrenal glands were hypertrophic; kidneys showed increased parenchymal echogenicity, loss of separation between the cortex and medulla, and enhanced medullary echogenicity; and size and echogenicity of the liver were normal. Magnetic resonance imaging of the abdomen confirmed that adrenal glands were hypertrophic (right and left adrenal gland were 24×22×18 mm and 18×19×20 mm in size, respectively) and lobulated. Echocardiogram revealed left ventricular hypertrophy. Bone survey verified generalized decrease in bone mass and revealed areas of irregular ossification and radiolucency in radius, ulna, and distal tibia, which were interpreted as osteoporosis due to hypercortisolism (Supplementary Figure S1).
Medical treatment
Metyrapone (300 mg/m2/day, per oral, in four doses) was started on the 25th day (Supplementary Figure S2) (6). Since liver function tests were impaired, metyrapone was preferred over ketoconazole. Soon after metyrapone was started, hyperglycemia and hypertension improved, enabling the discontinuation of insulin and nifedipine. Spironolactone was also gradually tapered and discontinued after 13 days of metyrapone treatment, and she was discharged.
The dose of metyrapone was adjusted frequently, according to clinical findings and serum cortisol levels during regular visits. However, even after gradually increasing metyrapone dose to 1,850 mg/m2/day over the course of 6 months, total biochemical suppression of serum cortisol could not be achieved (Supplementary Figure S3A), and the patient had progressive loss of bone mineral density, persistent left ventricular hypertrophy, and a lack of catch-up growth. In addition to that, café-au-lait macules became darker, dehydroepiandrosterone sulfate (DHEA-S) gradually increased (Table 2), and previously non-existent marked clitoromegaly was noted as a side effect of high-dose metyrapone. She was also prescribed ursodeoxycholic acid (15 mg/kg/day); however, liver enzymes remained high (Table 1).
Right total and left three-quarters adrenalectomy
Right total and left three-quarters adrenalectomy was carried out at 9 months of age in light of the patient’s continued clinical findings of hypercortisolism, the existence of unfavorable prognostic markers (high cortisol levels upon admission and heart and liver problems), and the adverse effects of high-dose metyrapone. The patient was administered 100 mg/m2/day glucocorticoids (GC) perioperatively; however, she developed symptoms of adrenal insufficiency. The required GC dose to attain euglycemia, restore general well-being, and resolve adrenal insufficiency was 300 mg/m2/day. Fludrocortisone (0.05 mg/day) was also started. Following surgery, supraphysiological doses of GC were required, as she suffered frequent symptoms of adrenal insufficiency (hypoglycemia, malaise, and loss of appetite). GC dose could be tapered very slowly, and a daily dose of 15 mg/m2/day could be attained in 2 months.
As liver function tests, serum cortisol levels and left ventricular hypertrophy all improved following adrenalectomy (Table 1). Bilateral nodular adrenal hyperplasia was observed in the pathological evaluation of surgical specimen, while the findings of liver wedge biopsy were non-specific (Supplementary Figure S4). Sequence analysis of GNAS from the surgical sample of adrenal gland revealed a heterozygous, previously described missense mutation in exon 8 (c.2530C>A, p.Arg844Ser), while the sequence analysis of the GNAS gene from peripheral blood sample was normal. Lymphocyte activation was normal 3 months post-adrenalectomy, and immunization schedule for live vaccines was established.
Other findings of MAS
She had breast development and vaginal bleeding that lasted 2 days when she was 7 months old, which repeated five more times after the adrenalectomy till 26 months of age. Breast development was Tanner stage 3, and bone age was markedly advanced (4 years and 2 months), despite severe hypercortisolism. On pelvic ultrasonography, uterus was enlarged to 34×22×24 mm; thus, letrozole (0.625 mg, per oral) was started at 26 months of age.
She also developed marked hypophosphatemia at the age of 6 months (Table 1). Radiological investigations since birth demonstrated severe osteopenia and lytic lesions, which were attributed to severe hypercortisolism; however, overt lesions of FD were not confirmed. When she was 9 months old, FGF-23 was elevated [122 pg/ml (normal <52)], which suggested hypophosphatemic rickets associated with FD. Oral phosphate (8 mg/kg) and calcitriol (18 ng/kg) were started. At the age of 23 months, bone survey revealed sclerosis of the base of the skull and maxilla and FD in the lower extremities. She has been on oral phosphate (58.7 mg/kg/day), while calcitriol was ceased.
She is now 34 months old with severe short stature [height, 81 cm (−3.5 SDS); weight, 9,580 g (−3.7SDS)] (Supplementary Figure S3B). She had been under regular clinic visits and has been on 15 mg/m2/day hydrocortisone and fludrocortisone 0.025 mg/day, letrozole (1×6.25 mg/day), phosphate (58 mg/kg), and ursodeoxycholic acid (100 mg/day) (Supplementary Figure S2). She has six words, cannot form two-word sentences, shows body parts, cannot stand up from supine position without support, and takes a few steps with support. Despite regular physiotherapy and ergotherapy, developmental delay is evident (Bayley Scales of Infant and Toddler Development III language scale, 13/79; motor scale, 2/46).
Discussion
ACTH-independent CS and café-au-lait macules suggested MAS in this case. Interestingly, this patient was admitted for hyponatremia and hyperglycemia requiring insulin treatment. Neonatal MAS and CS are rare conditions, and presentation of this case is quite unique (4).
The earlier the timing of somatic mutation, the greater the burden of Gsα-mutation-bearing cells leading to widespread tissue involvement in MAS. In the current case, adrenal, hepatic, cardiac, renal, and bone tissue involvement were evident in first weeks of life, while precocious puberty and hypophosphatemic rickets were observed later. A lifetime risk of additional tissue involvement is being acknowledged. CS is the rarest endocrine manifestation of MAS, which appears in <5%–7.1%. It presents exclusively within the first year of life (median age, 3.1 months) where features may develop as early as in utero (2–4, 7). The fact that our case was SGA and had moon facies and hirsutism with impaired linear growth, weight gain, hyperglycemia, hypertension, and nephrocalcinosis detected in the neonatal period, suggested severe, in utero onset CS. Upon suspicion, both comorbidities (hyperthyroidism, excess growth hormone, FD, and cardiac and hepatobiliary function) of MAS and complications of GC excess (hypertension, hyperglycemia, hyperlipidemia, nephrocalcinosis, decreased bone mineral density, and muscle atrophy) were assessed (1, 3).
Since the initial description of MAS, only 20 neonates with CS have been described with various initial basal serum cortisol ranging from 9.6 to 80.1 µg/dl, and data regarding long-term follow-up and outcome are still developing (1, 2, 8–11). Disease course is heterogenous, and spontaneous resolution of hypercortisolism has been reported (30%) since Gs-bearing cells are mostly located in the fetal adrenal zone, which normally undergoes apoptosis after birth. However, the outcome is mostly unfavorable in cases with extensive endocrine and extra-endocrine manifestations (1, 2, 8–15). Brown et al. reported poorer prognosis and a lower likelihood of spontaneous remission of adrenal disease in patients with cardiac (cardiomyopathy) and liver involvement (hepatocellular adenomas, inflammatory adenomas, choledochal cysts, neonatal cholestasis, and hepatoblastoma). It was hypothesized that these patients have a greater burden of Gsα mutation (3, 4).
Treatment of neonatal CS is a long and challenging path where both cortisol excess and its complications should be targeted. Marked hypercortisolism that precipitate neonatal diabetes requiring insulin treatment like our patient is rare and was previously reported only in six patients with CS (4). Until hypercortisolism is managed, hyperglycemia should be treated with insulin. Hypertension is due to mineralocorticoid effect of excess cortisol; thus, blood pressure lowering agents of choice should be aldosterone antagonists (spironolactone) or potassium-sparing diuretics.
The treatment strategy of hypercortisolism is determined by disease severity. In a mildly affected case, medical treatment with an expectation of spontaneous resolution (due to previously stated apoptosis of fetal adrenal zone) may be of choice (3, 4, 16–19). Metyrapone, ketoconazole, and mitotane are medical options for lowering cortisol (20–23). Since our patient had impaired liver function, metyrapone, a potent, rapid acting relatively selective inhibitor of 11-hydroxylase was preferred over ketoconazole for its low risk of hepatotoxicity. Reports reviewing adult data suggest an initial dose of 500–750 mg/day and achievement of biochemical control with 1,500 mg/day (23). However, the initial and maximum dose of metyrapone in neonates is unclear; some authors recommend 300 mg/m2/day in four equal doses (6). In our case, adequate biochemical and clinical suppression of cortisol with metyrapone was not achieved despite an increase in dose from 300 to 1,850 mg/m2/day.
There are important issues to be considered while using a steroidogenesis inhibitor like metyrapone. Monitoring biochemical response is essential, not only for dose titration and management of cortisol excess but also for adrenal insufficiency due to possible overtreatment. Clinical signs of adrenal insufficiency should always be questioned and assessed. The 24-h urinary free cortisol is the commonly used method; however, it may be impractical due to difficulties in the collection of urine in infants. Alternative methods may be the measurement of early morning serum cortisol and ACTH (23). Low ACTH level may indicate hypercortisolism or may be a sign of suppression due to long-term exposure to hypercortisolism. However, there are deadlocks to be considered in the evaluation of these measurements. A high cortisol level measured by immunoassays does not always indicate an actual elevation. It should be kept in mind that cortisol immunoassays exhibit significant cross-reactivity with cortisol precursors that may be elevated in patients treated with a steroidogenesis inhibitor (especially with metyrapone, which is known to increase 11-deoxycortisol). Such cross-reactivity can be a cause for overestimation of cortisol and may lead to risk of overtreatment (24, 25). It has been suggested that the patients on metyrapone should be biochemically monitored via specific methods, such as mass spectrometry (24–26).
Metyrapone is a relatively selective inhibitor of 11-hydroxylase and 18-hydroxylase. Recent in vitro studies indicate greater inhibitory action of metyrapone on aldosterone synthase, resulting in significant reversible reduction in both cortisol and aldosterone. The loss of negative feedback leads to an increase in ACTH, which causes an accumulation of cortisol and aldosterone precursors resulting in an increase in adrenal androgens (23). Although we could not serologically prove an increase in ACTH, hyperpigmentation and the increase in adrenal androgens confirm this mechanism. As far as we know, an increase in DHEA-S causing virilization was an unreported side effect of metyrapone. Clinical (clitoromegaly and hirsutism) and laboratory (DHEA-S) signs of hyperandrogenism should be monitored when higher doses of metyrapone are required.
In the severely affected case with CS, where medical treatment is inadequate and the chance of spontaneous resolution is subsiding, adrenalectomy is indicated when medically feasible. Brown et al. suggested that the presence of comorbid cardiac and liver disease like in our case should prompt consideration for early adrenalectomy (4). Although a previous correlation with initial serum cortisol level and prognosis was not established, it may be speculated that excessively high serum cortisol level is associated with increased number of Gsα-mutation-bearing adrenal cells. Thus, we suggest that in neonatal CS due to MAS, initial very high serum cortisol levels, like our case, may be a negative prognostic factor both for spontaneous resolution and clinical response to medical treatment. In infants with severe CS, bilateral adrenalectomy is generally performed. Alternatives like unilateral adrenalectomy and one-side total, other-side three-quarters adrenalectomy may be considered to avoid the requirement for lifelong GC and mineralocorticoid replacement. Unilateral adrenalectomy was reported to successfully improve clinical symptoms and endocrinological status in adult studies; nevertheless, recurrence during follow-up was 23.1%, while 17.5% required contralateral adrenalectomy (27–29). Since the causes of CS in adult series are variable and different from pediatric CS due to MAS, it should be borne in mind that reproducibility of adult data is poor. In CS due to MAS, Gsα-mutation-bearing adrenal gland cells are heterogeneously distributed, and partial adrenalectomy may carry the risk of inadequate management and recurrence. Only a few pediatric case reports addressed this issue. Unilateral adrenalectomy of the larger gland was performed in two neonates with CS due to MAS; remission was achieved for 2 years (30, 31). Itonaga et al. reported a 6-month-old neonate with MAS-associated CS treated with right-sided total adrenalectomy and left-sided half adrenalectomy with remission for 2 years (32). Although these cases were less severe [basal serum cortisol: 16.9, 18.5, and 23.4 µg/dl, respectively (N: 6.2–18.0 µg/dL)], we preferred to perform partial adrenalectomy (right total and left three-quarters adrenalectomy) and succeeded. Our patient has been in remission for more than 2 years.
In the largest case–control analysis of CS in patients with MAS, overall mortality was 20% (six cases) where four of them were deceased following bilateral adrenalectomy (66.7% of all deaths) (4). Anaphylaxis (or adrenal insufficiency), sudden cardiac arrest, sepsis, and sudden death were listed as causes of mortality in those four cases where GC dose and process of GC tapering were not clearly described. The fact that our patient required high-dose GC during peri- and postoperative period to restore well-being, tapering to maintenance dose was very slow, and she is still on maintenance dose GC, suggests that rapid tapering of GCs should be avoided and, although being speculative, may explain sudden death following adrenalectomy.
Gross motor developmental delay may be caused by prenatal exposure to excess GCs. Prenatal GC treatment for possible congenital adrenal hyperplasia or risk of premature birth have been shown to result in cognitive deficits after birth. Furthermore, children who develop CS later in life may experience a decline in cognitive and school performance where the younger the age of onset, the greater the deterioration in IQ scores (3, 4, 33, 34). Since transgenic mice with Gsα mutation was shown to have short- and long-term memory deficits and impaired associative and spatial learning, it may also be speculated that Gsα mutation may also be present in the central nervous system (35, 36).
The establishment of diagnosis of FD follows a characteristic and predictable time course. Although GNAS mutations are acquired early in embryogenesis, skeletal development appears to be relatively normal in utero, without frank clinical signs of FD at birth. Boyce et al. affirmed that FD lesions become apparent over the first several years of life and expand during childhood and adolescence, like our case. Previous case reports have also stated severe osteoporosis, rickets, polyostotic irregular lucencies, pathological fractures, and biopsy-proven FD during infancy (1, 2, 8–15). The exact pathophysiological mechanism is unclear, and Gsα activation in abnormally differentiated osteocytes is accused. FGF-23 overproduction is an inherent feature of FD, and most patients have elevated circulating levels of FGF-23, but frank hypophosphatemia is rare. The increase in FGF-23 is linked to substantial skeletal involvement. Although FGF-23 levels may wax and wane over time, an increase in FGF-23 usually occurs during periods of rapid growth like infancy and adolescence. Concurrent hyperfunctioning endocrinopathies like hyperthyroidism or CS may also adversely affect bone health.
Peripheral precocious puberty (PP) is the most frequent presenting feature in female patients with MAS (85%) (6). To date, a safe, effective, and long-term treatment for PP in girls with MAS has not been established. The benefits of current interventions on the ultimate outcome of interest, adult height, have not been well-established due to the rarity of the condition and heterogeneous nature of the disease. Despite the small sample size, studies have concluded that letrozole resulted in a statistically significant decrease in the bone age/chronological age ratio, growth velocity, hence increasing predicted adult height (37). Growth outcome in MAS is not only dependent on timing of pubertal onset but on several other disease components (skeletal involvement and endocrinopathies) as well. Hyperthyroidism and growth hormone excess may accelerate growth, while CS may decelerate it (37, 38).
Lack of consensus on both medical and surgical treatment strategies were major obstacles while navigating this case of severe neonatal MAS. The eminence of this report is that it presents current literature with clinical experience on this rare case of neonatal CS due to MAS. High index of suspicion for MAS in a neonate with extensive café-au-lait macules and symptoms of hypercortisolism is the key for early recognition and intervention. Initial excessive cortisol in neonatal CS may be a negative prognostic factor for spontaneous resolution and response to medical treatment, indicating early right total and left three-quarters adrenalectomy. Post-adrenalectomy survival may be related to close supervision during GC tapering.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
YU collected and analyzed data, drafted the initial manuscript, and reviewed and revised the manuscript. OG collected data. İU, HH, BG, SE, and TK collected data and reviewed and revised the manuscript. ZO and EG analyzed data, conceptualized the work, and revised and critically reviewed the manuscript for important intellectual and medical content. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Acknowledgments
We thank our patient’s family for providing consent for publication of this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1209189/full#supplementary-material
Supplementary Figure 1 | (A) The findings of physical and radiologic examination. Notice cushingoid facies, hyperpigmented macules that does not cross the midline at the front of the trunk. (B) Anteroposterior radiographs reveal irregularities in radius, ulna and femur. Although generalized osteopenia improves at 34 months, FD lesions become prominent over months.
Supplementary Figure 2 | Timeline of the course of symptoms in neonatal McCune Albright Syndrome noting adjustments made in treatment. Grey box denotes age in days for the first month of life then in months. NPH: Neutral Protamine Hagedorn insulin, CS: Cushing syndrome, PP: precocious puberty.
Supplementary Figure 3 | (A) Change in serum cortisol with increased metyrapone (methyrapone was initiated on day 25). (B) Growth chart, the arrow represents right total and left three quarters adrenalectomy.
Supplementary Figure 4 | Representative histological features of nodular adrenal hyperplasia. (A,
") show low-power while (C) Show high-power views.
show low-power while (C) Show high-power views.
References
1. Lourenço R, Dias P, Gouveia R, Sousa AB, Oliveira G. Neonatal McCune-Albright syndrome with systemic involvement: a case report. J Med Case Rep (2015) 9:189. doi: 10.1186/s13256-015-0689-2
2. Corsi A, Cherman N, Donaldson DL, Robey PG, Collins MT, Riminucci M. Neonatal McCune-Albright syndrome: A unique syndromic profile with an unfavorable outcome. JBMR Plus (2019) 3:e10134. doi: 10.1002/jbm4.10134
3. Boyce AM, Collins MT. Fibrous dysplasia/McCune-Albright syndrome: A rare, mosaic disease of Gα s activation. Endocr Rev (2020) 41(2):345–70. doi: 10.1210/endrev/bnz011
4. Brown RJ, Kelly MH, Collins MT. Cushing syndrome in the McCune-Albright syndrome. J Clin Endocrinol Metab (2010) 95(4):1508–15. doi: 10.1210/jc.2009-2321
5. Boyce AM, Florenzano P, de Castro LF, Collins MT. Fibrous Dysplasia/McCune-Albright Syndrome. Adam MP, Ardinger HH, Pagon RA, et al, editors. Seattle (WA): University of Washington, Seattle (2015).
6. Dias R, Storr HL, Perry LA, Isidori AM, Grossman AB, Savage MO. The discriminatory value of the low-dose dexamethasone suppression test in the investigation of paediatric Cushing's syndrome. Horm Res (2006) 65(3):159–62. doi: 10.1159/000091830
7. Carney JA, Young WF, Stratakis CA. Primary bimorphic adrenocortical disease: cause of hypercortisolism in McCune-Albright syn- drome. Am J Surg Pathol (2011) 35:1311–26. doi: 10.1097/PAS.0b013e31821ec4ce
8. Shenker A, Weinstein LS, Moran A, Pescovitz OH, Charest NJ, Boney CM, et al. Severe endocrine and nonendocrine manifestations of the McCune-Albright syndrome associated with activating mutations of stimulatory G protein GS. J Pediatr (1993) 123:509–18. doi: 10.1016/S0022-3476(05)80943-6
9. Danon M, Robboy SJ, Kim S, Scully R, Crawford JD. Cushing syndrome, sexual precocity, and polyostotic fibrous dysplasia (Albright syndrome) in infancy. J Pediatr (1975) 87:917–21. doi: 10.1016/S0022-3476(75)80905-X
10. Yoshimoto M, Nakayama M, Baba T, Uehara Y, Niikawa N, Ito M, et al. A case of neonatal McCune-Albright syndrome with Cushing syndrome and hyperthyroidism. Acta Paediatr Scand (1991) 80:984–7. doi: 10.1111/j.1651-2227.1991.tb11769.x
11. Kirk JM, Brain CE, Carson DJ, Hyde JC, Grant DB. Cushing’s syndrome caused by nodular adrenal hyperplasia in children with McCune- Albright syndrome. J Pediatr (1999) 134:789–92. doi: 10.1016/S0022-3476(99)70302-1
12. Lodish MB, Keil MF, Stratakis CA. Cushing's syndrome in pediatrics: an update. Endocrinol Metab Clin North Am (2018) 47(2):451–62. doi: 10.1016/j.ecl.2018.02.008
13. Post EM, Consenstein L, Hitch D, Oliphant M, Dracker R, Richman RA. Congenital Cushing syndrome with polyostotic fibrous dysplasia (PFD). Pediatr Res (1983) 17:169A.
14. Silva ES, Lumbroso S, Medina M, Gillerot Y, Sultan C, Sokal EM. Demonstration of McCune-Albright mutations in the liver of children with high gamma GT progressive cholestasis. J Hepatol (2000) 32:154–8. doi: 10.1016/S0168-8278(00)80202-0
15. Angelousi A, Fencl F, Faucz FR, Malikova J, Sumnik Z, Lebl J, et al. McCune Albright syndrome and bilateral adrenal hyperplasia: the GNAS mutation may only be present in adrenal tissue. Hormones (Athens) (2015) 14:447–50. doi: 10.14310/horm.2002.1578
16. Collins MT, Singer FR, Eugster E. McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet J Rare Dis (2012) 7. doi: 10.1186/1750-1172-7-S1-S4
17. Stratakis CA. Diagnosis and clinical genetics of Cushing syndrome in pediatrics. Endocrinol Metab Clin North Am (2016) 45(2):311–28. doi: 10.1016/j.ecl.2016.01.006
18. Bocian-Sobkowska J, Malendowicz LK, WoŸniak W. Comparative stereological study on zonation and cellular composition of adrenal glands of normal and anencephalic human fetuses. I. Zonation of the gland. Histol Histopathol (1997) 12:311–7.
19. Breault L, Chamoux E, Lehoux JG, Gallo-Payet N. Localization of G protein α-subunits in the human fetal adrenal gland. Endocrinology (2000) 141(12):4334–41. doi: 10.1210/endo.141.12.7834
20. Lake-Bakaar GSP, Sherlock S. Hepatic reactions associated with ketoconazole in the United Kingdom. BMJ (1987) 294:419–22. doi: 10.1136/bmj.294.6569.419
21. Heiberg JK, Svejgaard E. Toxic hepatitis during ketoconazole treatment. BMJ (1981) 283:825–6. doi: 10.1136/bmj.283.6295.825
22. Newell-Price J. Ketoconazole as an adrenal steroidogenesis inhibitor: Effectiveness and risks in the treatment of Cushing’s disease. J Clin Endocrinol Metab (2014) 99:1586–8. doi: 10.1210/jc.2014-1622
23. Daniel E, Newell-Price JD. Therapy of endocrine disease: steroidogenesis enzyme inhibitors in Cushing's syndrome. Eur J Endocrinol (2015) 172(6):R263–80. doi: 10.1530/EJE-14-1014
24. Owen LJ, Halsall DJ, Keevil BG. Cortisol measurement in patients receiving metyrapone therapy. Ann Clin Biochem (2010) 47:573–5. doi: 10.1258/acb.2010.010167
25. Monaghan PJ, Owen LJ, Trainer PJ, Brabant G, Keevil BG, Darby D. Comparison of serum cortisol measurement by immunoassay and liquid chromatography-tandem mass spectrometry in patients receiving the 11β-hydroxylase inhibitor metyrapone. Ann Clin Biochem (2011) 48:441–6. doi: 10.1258/acb.2011.011014
26. Monaghan PJ, Keevil BG Trainer PJ. The use of mass spectrometry to improve the diagnosis and the management of the HPA axis. Rev Endocrine Metab Disord (2013) 14:143–57. doi: 10.1007/s11154-013-9240-1
27. Li J, Yang CH. Diagnosis and treatment of adrenocorticotrophic hormone-independent macronodular adrenocortical hyperplasia: a report of 23 cases in a single center. Exp Ther Med (2015) 9:507–12. doi: 10.3892/etm.2014.2115
28. Debillon E, Velayoudom-Cephise FL, Salenave S, Caron P, Chaffanjon P, Wagner T, et al. Unilateral adrenalectomy as a first-line treatment of Cushing’s syndrome in patients with primary bilateral macronodular adrenal hyperplasia. J Clin En- docrinol Metab (2015) 100:4417–24. doi: 10.1210/jc.2015-2662
29. Albiger NM, Ceccato F, Zilio M, Barbot M, Occhi G, Rizzati S, et al. An analysis of different therapeutic options in patientswith Cushing’s syndrome due to bilateral macronodular adrenal hyperplasia: a single-centre experience. Clin Endocrinol (Oxf) (2015) 82:808–15. doi: 10.1111/cen.12763
30. Hamajima T, Maruwaka K, Homma K, Matsuo K, Fujieda K, Hasegawa T. Unilateral adrenalectomy can be an alternative therapy for infantile onset Cushing' s syndrome caused by ACTH-independent macronodular adrenal hyperplasia with McCune-Albright syndrome. Endocr J (2010) 57(9):819–24. doi: 10.1507/endocrj.K10E-003
31. Paris F, Philibert P, Lumbroso S, Servant N, Kalfa N, Sultan C. Isolated Cushing's syndrome: an unusual presentation of McCune-Albright syndrome in the neonatal period. Horm Res (2009) 72(5):315–9. doi: 10.1159/000245934
32. Itonaga T, Goto H, Toujigamori M, Ohno Y, Korematsu S, Izumi T, et al. Three-quarters adrenalectomy for infantile-onset cushing syndrome due to bilateral adrenal hyperplasia in McCune-Albright syndrome. Horm Res Paediatr (2017) 88(3-4):285–90. doi: 10.1159/000473878
33. Merke DP, Giedd JN, Keil MF, Mehlinger SL, Wiggs EA, Holzer S, et al. Children experience cognitive decline despite reversal of brain atrophy one year after resolution of Cushing syndrome. J Clin Endocrinol Metab (2005) 90(5):2531–6. doi: 10.1210/jc.2004-2488
34. Keil MF, Merke DP, Gandhi R, Wiggs EA, Obunse K, Stratakis CA. Quality of life in children and adolescents 1-year after cure of Cushing syndrome: a prospective study. Clin Endocrinol (Oxf) (2009) 71(3):326–33. doi: 10.1111/j.1365-2265.2008.03515.x
35. Bourtchouladze R, Patterson SL, Kelly MP, Kreibich A, Kandel ER, Abel T. Chronically increased Gsα signaling disrupts associative and spatial learning. Learn Mem (2006) 13:745–52. doi: 10.1101/lm.354106
36. Kelly MP, Cheung YF, Favilla C, Siegel SJ, Kanes SJ, Houslay MD, et al. Constitutive activation of the G-protein subunit Gαs within forebrain neurons causes PKA-dependent alterations in fear conditioning and cortical Arc mRNA expression. Learn Mem (2008) 15:75–83. doi: 10.1101/lm.723708
37. Feuillan P, Calis K, Hill S, Shawker T, Robey PG, Collins MT. Letrozole treatment of precocious puberty in girls with the McCune-Albright syndrome: a pilot study. J Clin Endocrinol Metab (2007) 92(6):2100–6. doi: 10.1210/jc.2006-2350
Keywords: McCune Albright syndrome, neonatal Cushing syndrome, metyrapone, adrenalectomy, follow-up
Citation: Unsal Y, Gozmen O, User İR, Hızarcıoglu H, Gulhan B, Ekinci S, Karagoz T, Ozon ZA and Gonc EN (2023) Case Report: Severe McCune–Albright syndrome presenting with neonatal Cushing syndrome: navigating through clinical obstacles. Front. Endocrinol. 14:1209189. doi: 10.3389/fendo.2023.1209189
Received: 20 April 2023; Accepted: 04 July 2023;
Published: 25 July 2023.Edited by:
Martin Oswald Savage, Queen Mary University of London, United KingdomReviewed by:
Li Chan, Queen Mary University of London, United Kingdom
Sasha R Howard, Queen Mary University of London, United Kingdom
Tomoyo Itonaga, Oita University, JapanCopyright © 2023 Unsal, Gozmen, User, Hızarcıoglu, Gulhan, Ekinci, Karagoz, Ozon and Gonc. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yagmur Unsal, yagmurunsal@yahoo.com
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
From https://www.frontiersin.org/articles/10.3389/fendo.2023.1209189/full
-
1
-
-
Abstract
Introduction and importance
Pheochromocytoma and Cushing's syndrome are rare endocrine conditions caused by tumors in the adrenal gland. These conditions are classified under Multiple Endocrine Neoplasia (MEN) syndrome, characterized by the development of multiple tumors in the endocrine system. However, diagnosing these conditions can be challenging as they often lack clear symptoms, requiring careful evaluation, monitoring, and treatment to prevent complications.
Case presentation
A 23-year-old male recently presented with right-sided abdominal fullness and lipoma-like masses on the torso. Over a span of six months, the abdominal mass nearly doubled in size, accompanied by elevated levels of catecholamines, cortisol, parathyroid hormone (PTH), and calcitonin. Surprisingly, the patient remained asymptomatic despite these abnormal lab values. CT imaging revealed a substantial increase in the size of the mass in the right adrenal gland, from 6 × 7 cm to approximately 11.2 × 10.2 × 9 cm.
Clinical discussion
Pheochromocytoma secretes catecholamines and often leads to hypertension and related symptoms. Interestingly, most individuals with pheochromocytoma do not exhibit obvious symptoms, necessitating blood and urine tests, along with imaging studies, for accurate diagnosis. The size of the tumor does not necessarily indicate the severity of symptoms. MEN-2, a genetic syndrome, is characterized by pheochromocytoma, medullary thyroid carcinoma, and hyperparathyroidism. Additionally, methods for diagnosing Cushing's syndrome, caused by excess cortisol production, are discussed.
Conclusion
Early diagnosis and genetic counseling are crucial in preventing complications associated with these conditions. By identifying them, appropriate treatment can be ensured for positive outcomes of patients and their families.
Keywords
PheochromocytomaMultiple Endocrine Neoplasia (MEN) syndromeCushing's syndromeRare Case ReportAbbreviations
- CT
- MRI
-
Magnetic resonance imaging
- USG
- 131I-MIBG
-
iodine 131 labeled meta-iodobenzylganidine
- RAAS
-
Renin-angiotensin-aldosterone system
1. Introduction
Pheochromocytoma are catecholamine secreting tumors of chromaffin cells of adrenal medulla. It can be found anywhere in the body, with the majority being intra-abdominal and those other than adrenal medulla are referred to as paragangliomas [1,2]. Pheochromocytoma typically secretes norepinephrine and epinephrine, with norepinephrine being the primary catecholamine. However, some tumors may only secrete one of the two, and rarely, some may secrete dopamine or dopa [3].
Vast majority >90 % of adrenal neoplasms are benign non-functional adenomas [4].About 10 % of pheochromocytomas are malignant and 10 % of cases are found on both sides. Additionally, approximately 40 % of pheochromocytomas are caused by genetic factors and can be associated with inherited syndromes [5].
Pheochromocytoma is found to be associated with MEN-2. MEN-2 is a hereditary genetic condition that is caused by a de novo mutation in the RET gene. It is inherited in an autosomal dominant fashion and is mainly characterized by medullary thyroid carcinoma, pheochromocytoma and parathyroid adenoma or hyperplasia [6].
MEN syndrome can be MEN-1, MEN-2A and MEN-2B. MEN-1 is characterized by pituitary tumors (prolactin or growth hormone), pancreatic endocrine tumors and parathyroid adenomas. Additionally, other tumors such as foregut carcinoids, adrenocortical adenomas, meningioma, lipomas, angiofibromas and collagenomas may also occur in MEN-1. MEN-2A is characterized by medullary thyroid carcinoma, pheochromocytoma, and parathyroid adenoma/hyperplasia; it can also be associated with cutaneous lichen amyloidosis and Hirschsprung disease. On the other hand, MEN-2B is characterized by familial medullary thyroid cancer, pheochromocytoma, mucosal neuromas, gastrointestinal tract issues, musculoskeletal and spinal problems. [7].
Cushing syndrome results from hypercortisolism and is characterized by hypertension, weight gain, easy bruising, and central obesity [4]. Cushing's disease refers to ACTH-dependent cortisol excess caused by a pituitary adenoma, while ACTH-independent cortisol excess due to non-pituitary causes such as excess use of glucocorticoids, adrenal adenoma, hyperplasia, or carcinoma is referred to as Cushing syndrome [8].
This case report has been written according to the SCARE checklist [9].
2. Case presentation
A 23-year-old male presented to our surgery department with the chief complaint of right sided abdominal fullness for six months. According to the patient a mass was incidentally reported six months back while he was under-evaluation for mild trauma due to road traffic accident. Six months back, the mass was approximately 6 × 7 cm, while at the time of presentation to our department the mass was approximately 11.2 × 10.2 × 9 cm (CT abdomen) which was globular in shape, had regular margin, and moved with respiration. He had no history of hypertension, headache, palpitation, sweating, pallor, recent weight loss, abdominal pain, psychological disturbance, dizziness, loss of consciousness, dark color urine, burning micturition, had normal bowel and bladder habit.
Past history and family history were insignificant. He was not under any long-term medication and no known drug allergies. He occasionally smokes and consumes alcohol.
On physical examination at the time of presentation, multiple soft, mobile, painless, subcutaneous nodules like lipoma were present over the torso. His height was 176.8 cm, weight 68 kg, BMI 21.8 kg/m2 (body mass index). He had blood pressure of 110/70 mm of Hg taken in left arm at sitting position, heart rate of 62 beats/min, respiratory rate of 24/min, temperature of 96.6 °F, SPO2 of 98 % at right hand. A mass was palpable on the right side of abdomen, otherwise abdomen was soft, non-tender, normal bowel sound was present. Chest, cardiac and neurologic examinations were all normal.
Initial laboratory evaluation revealed 24 h. urine metanephrine of 5415 μg/24 h (normal: 25–312 μg/24 h.); 24 h. urine VMA of 32.2 mg/24 h. (normal: <13.60 mg/24 h.); serum cortisol of 535.16 nmol/l after overnight low dose dexamethasone(1 mg) suppression test (normal: <50 nmol/l);24 h. Urine free cortisol of 526.61 nmol/24 h. (normal: 30–145 nmol/24 h) PTH(intact) of 89.2 pg./ml (normal: 15–65 pg./ml); serum calcitonin of 15.2 pg./ml (normal: ≤8.4 pg./ml); serum CEA of 4.72 ng/ml (normal: 0.0–4.4 ng/ml); serum DHEA of 1.19 ng/ml (normal: 1.7–6.1 ng/ml). Baseline investigation: Hematology, urine routine/microscopic, electrolytes were within the normal range.
Additional laboratory findings were as in the Table 1.
Table 1.
Lab evaluation Result Reference Unit Metanephrine, urine 24 h 5415 25–312 μg/24 h VMA, urine 24 h 32.2 <13.60 mg/24 h VMA, urine 12.88 – ng/l Cortisol, serum, overnight DST 535.16 <50 nmol/l Cortisol, urine 24 h 526.61 30–145 nmol/24 h ACTH, complete 28.3 7.2–63.3 pg/ml DHEA, serum 1.19 1.7–6.1 ng/ml CEA, serum 4.72 0.0–4.4 ng/ml Phosphorus, serum 3.0 2.5–4.5 mg/dl Albumin, serum 5.2 3.5–5.2 g/dl Calcitonin, serum 15.2 ≤8.4 pg/ml Calcium, serum 8.94 8.6–10.0 mg/dl PTH (intact) 89.2 15–65 pg/ml aldosterone 8.7 7.0–30 g/dl Plasma rennin activity 1.42 0.10–6.56 ng/ml/h Aldosterone-rennin ratio 6.13 ≤20 Creatinine, urine 36 – mg/dl DST - dexamethasone suppression test; VMA - vanilmandelic acid; ACTH - adrenocorticotropic hormone; DHEA - dehydroepiandrosterone; CEA - carcino-embryonic-antigen; PTH - parathyroid hormone.
2.1. USG abdomen
USG abdomen (Fig. 1, Fig. 2) showed well defined mixed echoic area in Right adrenal region measuring 12.7 × 10.7 cm in size. There was presence of internal vascularity with multiple foci of cystic compound. The lesion displaced the right kidney inferiorly.

Fig. 1. USG abdomen.

Fig. 2. USG abdomen.
2.2. Plane and contrast CT scan of abdomen
Plane and contrast CT scan of Abdomen (Fig. 3) showed approximately 11.2 × 10.2 × 9 cm sized, relatively well defined heterogeneous soft tissue density lesion with well-defined enhancing wall in right adrenal region. Non-enhancing areas were noted within the mass suggestive of necrosis. Few calcific foci were noted within the mass with no obvious hemorrhagic component. The lesion showed heterogeneous enhancement post contrast image.

Fig. 3. CT abdomen.
After all the workup patient was given diagnosis of right sided Pheochromocytoma associated with MEN syndrome, with ACTH-independent Cushing's syndrome and right adrenalectomy was performed.
2.3. Pathology report
2.3.1. Gross descriptions
The specimen was globular mass measuring 14.5 × 10 cm, with smooth outer surface. On sectioning, the mass was well circumscribed, soft and yellow-brown, predominantly solid with cyst formation. The size of cyst ranges from 0.3 to 3.5 cm in diameter. Areas of hemorrhages were noted.
2.3.2. Microscopic description
Section showed tumor cells arranged in well-defined nests (Zellballen), alveolar and diffuse pattern with intervening fibrovascular stroma. The cells were intermediate to large sized, polygonal with finely granular amphophilic cytoplasm. The nuclei showed mild to moderate pleomorphism and were round to ovoid, with prominent nuclei noted. No capsular invasion, vascular invasion and necrosis. Areas of hemorrhage were seen. Mitosis 0–1/10 high power field was noted (Figs. 4 and 5).

Fig.a Diffuse Zellbalen pattern with intervening fibrous stroma.
Fig.b Mild to moderate pleomorphic nuclei with abundant hemorrhage.
Fig.c Low power field with intact capsule.

Figs. 4 and 5. Fig. 4 Intra-operative resection of tumor; Fig. 5 tumor after resection.
3. Discussion
In Pheochromocytoma activation of the alpha-one adrenergic receptor by catecholamine in the vascular bed causes vasoconstriction and leads to a rise in blood pressure. Similarly, activation of the beta-one receptor in the heart enhances the chronotropic and inotropic effect of the myocardium, leading to an increase in heart rate and cardiac output. In addition, activation of the beta-one receptor in the juxtaglomerular cells of the kidney activates the RAAS system. These receptor activation result in cardiovascular and sympathetic changes, such as hypertension, palpitation, headache, sweating, trembling, and anxiety [10].
In Pheochromocytoma, the patient may have a 10-fold increase in plasma catecholamines, but the hemodynamic response can still fall within the normal range due to desensitization of the cardiovascular system. When catecholamine levels are elevated for a prolonged period, the alpha-one receptors in blood vessels may be down-regulated, making norepinephrine unresponsive in raising peripheral vascular resistance, which can lead to normal blood pressure. Similarly, a marked decrease in beta-one receptors in the heart could explain the normal heart rate, which was observed in our asymptomatic patient with Pheochromocytoma [11].
Sometimes in asymptomatic patients, the size of the tumor tends to be larger than in those with hyperfunctioning tumors [12]. However, medical interventions such as surgery, anesthesia induction, intravenous urography contrast, or manipulation of the tumor can trigger adrenergic and hypertensive crises, so biopsy is usually contraindicated in pheochromocytoma [13].
The diagnosis of pheochromocytoma is typically based on measuring plasma and urinary levels of catecholamines and their derivatives such as metanephrine and vanillylmandelic acid. The most reliable test is the measurement of urinary metanephrine as its excretion levels are relatively higher [13,14]. The combination of 131I-MIBG scintigraphy along with diagnostic urinary and blood tests can further enhance the sensitivity of the test. Specifically, the urinary normetanephrine test is considered the most sensitive single test for detecting Pheochromocytoma [15,16].
In addition to a 24-h urine test and blood test, if the lab results are positive for Pheochromocytoma or paragangliomas, further diagnostic tests may be recommended, such as a CT scan, MRI, m-iodobenzylganidine (MIBG) imaging, or positron emission tomography (PET) [16,17]. In our patient 24 h. urine metanephrine of 5415 μg/24 h (normal: 25–312 μg/24 h.); 24 h. urine VMA of 32.2 mg/24 h. (normal: <13.60 mg/24 h.) and imaging confirmation of right adrenal mass lead to the diagnosis of right sided pheochromocytoma.
Our patient with pheochromocytoma was tested for parathyroid hormone and calcitonin due to the association of pheochromocytoma with MEN-2 [18]. MEN-2 can be diagnosed biochemically by measuring the baseline levels of calcitonin, parathyroid hormone and serum calcium along with blood tests for catecholamines and their metabolites to detect pheochromocytoma [19]. In our patient, multiple soft, mobile, painless, subcutaneous nodules like lipoma were present over the torso(MEN-1) and high levels of parathyroid hormone and calcitonin were detected(MEN-2). These findings can be correlated with MEN syndrome.
USG of the neck revealed no abnormalities of thyroid and parathyroid gland in our patient so prophylactic thyroidectomy was not done, instead he was counseled for follow up if any symptoms or thyroid swelling appears.
The diagnosis of Cushing's syndrome typically involves measuring the levels of 24-h urine free cortisol and assessing the suppression of cortisol in response to a 1 mg overnight dexamethasone test. If cortisol levels remain elevated despite the test, the next step is to measure serum ACTH levels. If ACTH levels are suppressed, it suggests an ACTH-independent cause of Cushing's syndrome, while elevated ACTH levels suggest an ACTH-dependent cause. Further evaluation may include a CT scan of the chest, abdomen, and pelvis to identify potential ectopic sources, as well as an MRI of the pituitary gland [8]. Our patient had a high level of 24 h. urine free cortisol of 526.61 nmol/24 h (reference range: 30–145 nmol/24 h) and serum cortisol of 535.16 nmol/L(reference range: <50 nmol/L) after overnight 1 mg dexamethasone suppression test, but normal level of ACTH of 28.3 pg./ml (reference range: 7.2–63.1 ng/ml), this suggests the diagnosis of ACTH independent Cushing's syndrome.
4. Conclusion
Large Pheochromocytoma patients can be asymptomatic and can present in association with other endocrine disorders. So proper evaluation is necessary to find out associated conditions and manage accordingly to prevent the possible outcomes.
Patient consent
Written, informed consent was obtained from the patient for the publication of the report.
Ethical approval
It is exempted at my institution. We don't need to take approval from ethical committee for case report.
Funding
N/A.
Author contribution
Conceptualization: Sanjit Kumar Shah.
Clinical diagnosis and patient management: Mahipendra Tiwari.
Microscopic slide preparation: Sneh Acharya.
Writing original draft: Sanjit Kumar Shah and Avish Shah.
All authors were involved in reviewing, editing, supervision and in preparing the final
manuscript.
Guarantor
Guarantor: Sanjit Kumar Shah
Email: sanjitshah023@gmail.com
Conflict of interest statement
N/A.
References
-
[1]
P.J. Klingler, T.P. Fox, D.M. Menke, J.M. Knudsen, J.T. FulmerPheochromocytoma in an incidentally discovered asymptomatic cystic adrenal massMayo Clin. Proc., 75 (5) (2000), pp. 517-520, 10.4065/75.5.517
-
[2]
K. Salmenkivi, J. Arola, R. Voutilainen, et al.Inhibin/activin betaB-subunit expression in pheochromocytomas favors benign diagnosisJ. Clin. Endocrinol. Metab., 86 (5) (2001), pp. 2231-2235, 10.1210/jcem.86.5.7446
-
[3]
W.M. Manger, R.W. GiffordPheochromocytomaJ. Clin. Hypertens (Greenwich)., 4 (1) (2002), pp. 62-72, 10.1111/j.1524-6175.2002.01452.xView PDFGoogle Scholar
This article is free to access.
-
[4]
S.R. Pingle, F. Jalil, D. Millar, C.D. Malchoff, B.T. RistauIsolated DHEAS production by an adrenal neoplasm: clinical, biochemical and pathologic characteristicsUrol. Case Rep., 31 (2020), Article 101148Published 2020 Feb 26Published 2020 Feb 26
-
[5]
I. Jandou, A. Moataz, M. Dakir, A. Debbagh, R. AboutaiebMalignant pheochromocytoma: a diagnostic and therapeutic dilemmaInt. J. Surg. Case Rep., 83 (2021), Article 106009, 10.1016/j.ijscr.2021.106009
-
[6]
L. Canu, G. Parenti, G. De Filpo, M. MannelliPheochromocytomas and Paragangliomas as causes of endocrine hypertensionFront. Endocrinol. (Lausanne)., 10 (2019), p. 333Published 2019 Jun 4Published 2019 Jun 4
-
[7]
G.G. Callender, T.A. Rich, N.D. PerrierMultiple endocrine neoplasia syndromesSurg. Clin. North Am., 88 (4) (2008), p. 863viiiviii
-
[8]
R. Pivonello, M. De Leo, A. Cozzolino, A. ColaoThe treatment of Cushing’s diseaseEndocr. Rev., 36 (4) (2015), pp. 385-486, 10.1210/er.2013-1048
-
[9]
R.A. Agha, T. Franchi, C. Sohrab, G. Mathew, A. Kirwan, A. Thomas, et al.The SCARE 2020 guideline: updating consensus Surgical Case Report (SCARE) guidelinesInt. J. Surg., 84 (1) (2020), pp. 226-230
-
[10]
L. Canu, G. Parenti, G. De Filpo, M. MannelliPheochromocytomas and Paragangliomas as causes of endocrine hypertensionFront. Endocrinol. (Lausanne)., 10 (2019), p. 333Published 2019 Jun 4Published 2019 Jun 4
-
[11]
E. Bravo, F. Fouad-Tarazi, G. Rossi, et al.A reevaluation of the hemodynamics of pheochromocytomaHypertension., 15 (2 Suppl) (1990), pp. I128-I131, 10.1161/01.hyp.15.2_suppl.i128
-
[12]
M.A. Blake, S.K. Krishnamoorthy, G.W. Boland, et al.Low-density pheochromocytoma on CT: a mimicker of adrenal adenomaAJR Am. J. Roentgenol., 181 (6) (2003), pp. 1663-1668, 10.2214/ajr.181.6.1811663
-
[13]
R. Yu, A. Pitts, M. WeiSmall pheochromocytomas: significance, diagnosis, and outcomeJ. Clin. Hypertens (Greenwich)., 14 (5) (2012), pp. 307-315, 10.1111/j.1751-7176.2012.00604.xView PDFView in ScopusGoogle Scholar
This article is free to access.
-
[14]
F. Zoghbi, C. Landault, N. Salmon, J.C. LegrandLes catécholamines et leurs dérivés dans l’exploration du phéochromocytome [Catecholamines and their derivatives in the study of pheochromocytoma]Ann. Med. Interne (Paris)., 134 (3) (1983), pp. 230-232
-
[15]
U. Guller, J. Turek, S. Eubanks, E.R. Delong, D. Oertli, J.M. FeldmanDetecting pheochromocytoma: defining the most sensitive testAnn. Surg., 243 (1) (2006), pp. 102-107, 10.1097/01.sla.0000193833.51108.24
-
[16]
G. Eisenhofer, M. PeitzschLaboratory evaluation of pheochromocytoma and paragangliomaClin. Chem., 60 (12) (2014), pp. 1486-1499, 10.1373/clinchem.2014.224832View PDFView in ScopusGoogle Scholar
This article is free to access.
-
[17]
H.P. Neumann, D.P. Berger, G. Sigmund, et al.Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel-Lindau disease [published correction appears in N Engl J Med 1994 Dec 1;331(22):1535]N. Engl. J. Med., 329 (21) (1993), pp. 1531-1538, 10.1056/NEJM199311183292103
-
[18]
V. Amodru, D. Taieb, C. Guerin, et al.MEN2-related pheochromocytoma: current state of knowledge, specific characteristics in MEN2B, and perspectives [published correction appears in endocrine. 2020 Jun 2;:] [published correction appears in endocrine. 2020 Jul 14;:]Endocrine, 69 (3) (2020), pp. 496-503, 10.1007/s12020-020-02332-2
-
[19]
C.J. Lips, J.W. Höppener, B.P. Van Nesselrooij, R.B. Van der LuijtCounselling in multiple endocrine neoplasia syndromes: from individual experience to general guidelinesJ. Intern. Med., 257 (1) (2005), pp. 69-77, 10.1111/j.1365-2796.2004.01429.xView PDFView in ScopusGoogle Scholar
This article is free to access.
-
Cushing's syndrome (CS) is a rare disease with multiple somatic signs and a high prevalence of co-occurring depression. However, the characteristics of depression secondary to CS and the differences from major depression have not been described in detail. In this case, we report a 17-year-old girl with treatment-resistant depression with a series of atypical features and acute psychotic episodes, which is a rare condition secondary to CS. This case showed a more detailed profile of depression secondary to CS and highlighted the differences with major depression in clinical features, and it will improve insight into the differential diagnosis especially when the symptoms are not typical.
Introduction
Depression is a chronic medical problem with typical features, including sadness, decreased interest and cognitive impairments. In clinical practice, depression can occur in other medical conditions, especially endocrinopathies, making it a more complex problem and exhibiting a challenge in diagnosis, especially in first-contact patients or when the clinical presentations are atypical. It is generally accepted that patients who failed to respond to two or more adequate trials of first-line antidepressants for treatment of major depressive episode are considered to have treatment-resistant depression (TRD) (1). For patients with TRD, a throughout evaluation should be performed to investigate the underlying organic causes.
Cushing's syndrome is a rare but serious endocrine disease due to chronic exposure to excess circulating glucocorticoids with multisystem effects (2). The etiology of CS can be divided into adrenocorticotropic hormone (ACTH)-dependent and ACTH-independent. It is characterized by a series of clinical features suggesting hypercortisolism, for example, metabolic abnormalities, hypertension and bone damages (3). A variety of neuropsychiatric symptoms, such as mood disturbance, cognitive impairment and psychosis, also occur in more than 70% CS patients (4). CS is life-threating if not timely diagnosed and treated, however, correct diagnosis can be delayed due to the wide range of phenotypes, especially when they are not classical (5).
Previous studies suggested that major depression was the most common co-morbid complication in CS patients, with a prevalence of 50–81% (6). Haskett's study confirmed that 80% of subjects meet the criteria for major depression with melancholic features (7). As reported in most recent investigations, depression in CS was not qualitatively different from non-endocrine major depression and the similarity was even striking (3, 8). However, some studies showed different conclusions and suggested a high prevalence of atypical depressive features other than melancholic features in CS (9). TRD and anxious depression has also been reported in CS patients (10, 11). All of the above conclusions suggest the complexity of depression with CS, and no distinct features have been found pertaining to hypercortisolism (12, 13). Although the intensity of depression secondary to CS is severe, suicidal depression is still an unusual condition (14).
Psychosis is a rare manifestation of CS, and the literature is limited. Only a few cases have been reported so far, especially combined with depression episode. In this case report, we presented a girl with CS, who experienced suicidal depression with a series of atypical features and acute psychotic symptoms, which was rarely reported in previous studies.
Case description
A 17-year-old girl with major depression for 3 years was involuntarily admitted for severe depressed mood with suicide attempts (neck cutting; tranquilizer overdose) and paranoid state in the last 2 weeks without any precipitating factors.
She experienced depressed and irritable mood in the last 3 years, and her condition had not improved although several adequate trials of antidepressants were used with satisfactory compliance (sertraline 200 mg/d; escitalopram oxalate 20 mg/d). Over the 2 weeks prior to admission, her depression continued to worsen with increasing irritability, she committed several suicide attempts, and once stated that she was unsafe at home. On admission, her heart rate was 116 bpm with blood pressure 139/81 mmHg and normal temperature; physical examination showed a cushingoid and virilising appearance (central obesity, swollen and hirsute face with acne, purple striae on the abdomen and bruises on the arms). No other abnormal signs were noted. She seemed drowsy but arousable, and she walked slowly, with bent shoulders and an inclined head. Mental state examination was hard to continue because she was passive and reluctant to answer our questions. Venlafaxine 150 mg/d has been used for more than 3 months with poor effects at that time.
Besides, weight gain (25 kg), irregular menstrual cycles and numbness of the hands and feet in the last half year were reported by her parents. Otherwise, No episodes of elevated mood and hyperactivity were found during the history taking. She does not have remarkable family history of serious physical or psychiatric illness; she was healthy, had an extroverted personality and had never used substances. Her premorbid social function and academic performance were good.
Several clinical characteristics found during the following mental state examinations were listed as follows:
• Prominent cognitive impairment without clouding of consciousness: Forgetfulness was frequently noted; she easily forgot important personal information such as her school and grade; she could not recall the suicide attempt committed recently and perfunctorily ascribed it to a casual event; and it was hard for her to recall her medical history (as it is for other depressive patients). The serial seven subtraction task could not be finished, and the interpretation of the proverb was superficial. Difficulty was found in attention maintenance; an effective conversation was hard to perform because she was mind-wandering (we needed to call her name to get her immediate attention) and often interrupted our conversations by introducing irrelevant topics or leaving without apparent reasons.
• Decreased language function that did not match her educational background: The patient could not find the proper words to articulate her feelings; instead, many simple, obscure and contradictory words were used, which made her response seem perfunctory. For example, she responded with “I do not know,” “I forgot,” or kept silent in response to our questions, which made the conversations hard to perform.
• Psychotic outbursts: Once she left the psychological therapy group, ranted about being persecuted and shook in fearfulness, stated “call the police” repeatedly, negative of explanations and comforts from others, but she cannot give any explanation about her behavior when calmed down. Sometimes she worried about being killed by the doctors but the worries were transient and fleeting.
• Depressed mood and negative thoughts (self-blame, worthlessness, and hopelessness) that were not persistent and profound: During most of her hospitalization, the patient seemed confused and apathetic, with intermittent anxiety, but she could not clearly express what made her anxious. Her crying and sadness happened suddenly, without obvious reasons, and she even denied low mood sometimes and said she had come to the hospital for cardiac disease treatment (she did not have any cardiac disease). Her description of her depressed mood was uncertain when specifically questioned, and she rarely reported her depressed feeling spontaneously as other depressed patients would. She did not even have the desire to get rid of her “depression”. Her suicidal ideation was transient and impulsive, and she could not provide a comprehensive explanation for her suicide attempts, such as emptiness, worthlessness or guilt. She was impatient and restless when interacting with others or when a more in-depth conversation was performed. She seemed apathetic, gave little response to emotional support from others and did not care about relevant important issues, such as hospital discharge or future plans. Elevated mood and motor activity were not found during the admission period.
• Social withdrawal and inappropriate behaviors: The patient often walked or stayed alone for long periods of time before speaking to other patients suddenly, which seemed improper or even odd in normal social interactions. During most hospitalization periods, lethargy and withdrawal were obvious.
Diagnostic assessment and therapeutic interventions
Basic laboratory tests reported abnormal results (Table 1), and the circulating cortisol level was far beyond the upper limit of normal, with a loss of circadian rhythm (Table 2); 24-h urinary free cortisol : >2897 nmol/24 h↑(69–345 nmol/24 h); serum ACTH (8 AM, 4 PM, 12 PM): 1.2 pg/ml, 1.3 pg/ml, <1 pg/ml (normal range: 1–46 pg/ml); low-dose dexamethasone suppression test (1 mg) (cortisol value): 1010.1 nmol/l (not suppressed; normal range: <50 nmol/L); high dose dexamethasone inhibition test (cortisol value): 879.0 nmol/l (not suppressed); OGTT and glycosylated hemoglobin; both normal. Other results used to rule out hyperaldosteronism and pheochromocytoma, such as the aldosterone/renin rate (ARR) and the vanillylmandelic acid, dopamine, norepinephrine and epinephrine levels, were reported to be within normal limits; ECG suggested sinus tachycardia; dual-energy X-ray bone density screening values were lower than the normal range; B-mode ultrasound showed a right adrenal tumor and fatty liver. The abdominal CT scan showed a tumor in her right adrenal gland. Brain MRI showed no abnormalities. Psychometric tests including HAMD (Hamilton depression scale), MADRS (Montgomery-Asberg Depression Rating Scale), WAIS (Wechsler Intelligence Scale) and MMSE (Mini-mental State Examination) were hard to perform due to her poor attention and non-cooperation presentation.
Table 1
Table 1. Abnormal lab results for the patient.
Table 2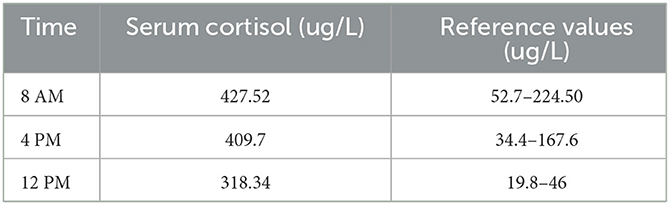
Table 2. Circulating cortisol level.
The patient had little response to adequate antidepressants in our hospital, including fluoxetine 20–60 mg/d and aripiprazole 5–30 mg/d combined with 3 sessions of MECT (modified electroconvulsive therapy), which was stopped because of her poor cognitive function and poor response.
Her last diagnosis was right adrenal adenoma and non-ACTH-dependent Cushing's syndrome. The adrenal adenoma was excised through laparoscopic resection in a general hospital. Hydrocortisone, amlodipine besylate, potassium chloride, metoprolol and escitalopram were used for treatment. Escitalopram 10 mg/d has been used until 2 weeks after her discharge. At the follow-up visit about 1 month after the surgery, her depressive mood had significantly improved, with no self-injury behaviors or psychiatric symptoms found. The patient was calm but still reacted slowly, and cognitive impairment was still found at the last visit.
Discussion
Previous studies have reported a close association between CS and depression (15). However, suicidal depression with atypical features and acute psychosis have rarely been reported, and the characteristics of depression secondary to CS and the differences from major depression have not been described in detail.
This case did not show a full-blown presentation of major depression according to the DSM-5. She presented with a series of features that were not typical as major depression, however, it should be emphasized that the atypical features were not identical to those noted in DSM5, especially regarding increased appetite and hypersomnia. The features suggesting difference from major depression were listed as follows: (a) depressed mood is not constant, it does not exist in most of the day; it is episodic without regular cyclicity, can happen or exacerbate suddenly; (b) the ability to describe anhedonia is poor, she can't report her feeling voluntarily like other patients with major depression, which might be partially related with the decreased language function; (c) depressive thoughts such as self-accusation and feelings of guilt, the classical symptoms of major depression, were rarely found; (d) more exaggerated cognitive impairment and decrease language function; € partial or little useful effect of SSRIs (selective serotonin reuptake inhibitors). The above characteristics were similar to those reported in Starkman's research (13, 16, 17), in which increasing irritability was also regarded as one of the important features for depression in CS.
The literature about depression combined with psychosis episode in CS is rare. This patient showed acute episodes of persecutory delusion with disturbed behaviors; her psychotic symptoms occurred suddenly and were fragmentary, with poor sensitivity to antipsychotics; the content was not constant (she never referred to and even denied the unsafe feeling at home before admission), it changed with the environment and was not consistent with the mood state. However, we cannot reach an effective conclusion because the evidence was small; thus, these findings should be evaluated in combination with other clinical presentations.
Conclusion
Most reviews have concluded that mood disturbances in CS indicate “major depression”, but the detailed description of clinical features are lack, making clinicians uncertain about the presentation and confused about the diagnosis, especially when the somatic signs are indiscriminate. The clinical presentation in this case highlighted the fact that there is a wide range of phenotypes of depression in CS, for some CS patients, the depressive features are not highly consistent with the criteria of major depression regardless of the melancholic or atypical features in the DSM-5. Thus, a thorough and periodic evaluation is necessary to detect the underlying organic and psychosocial causes if the clinical symptoms are not typical (10).
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
XY, SC, XJ, and XH were responsible for clinical care. XY did literature search and drafted the manuscript. XH revised the manuscript. All authors contributed to the article and have approved the final manuscript.
Acknowledgments
We want to thank Juping Fu, Ying Zhang, and all other medical staff who gave careful nursing to the patient.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kverno KS, Mangano E. Treatment-resistant depression: approaches to treatment. J Psychosoc Nurs Ment Health Serv. (2021) 59:7–11. doi: 10.3928/02793695-20210816-01
2. Mokta J, Sharma R, Mokta K, Ranjan A, Panda P, Joshi I. Cushing's disease presenting as suicidal depression. J Assoc Physicians India. (2016) 64:82–3.
3. Sonino N, Fava G. A Psychiatric disorders associated with Cushing's syndrome epidemiology, pathophysiology and treatment. CNS Drugs. (2001) 15:361–73. doi: 10.2165/00023210-200115050-00003
4. Pivonello R, Simeoli C, De Martino MC, Cozzolino A, De Leo M, Iacuaniello D, et al. Neuropsychiatric disorders in Cushing's syndrome. Front Neurosci. (2015) 9: 129. doi: 10.3389/fnins.2015.00129
5. Barbot M, Zilio M, Scaroni C. Cushing's syndrome: Overview of clinical presentation, diagnostic tools and complications. Best Pract Res Clin Endocrinol Metab. (2020) 34:101380. doi: 10.1016/j.beem.2020.101380
6. Sonino N, Fava GA, Raffi AR, Boscaro, Fallo F. Clinical correlates of major depression in Cushing's disease. Psychopathology. (1998) 31:302–6. doi: 10.1159/000029054
7. Haskett RF. Diagnostic categorization of psychiatric disturbance in Cushing's syndrome. Am J Psychiatry. (1985) 142:911–6. doi: 10.1176/ajp.142.8.911
8. Fava GA. Affective disorders and endocrine disease. New insights from psychosomatic studies. Psychosomatics. (1994) 35:341–53. doi: 10.1016/S0033-3182(94)71755-2
9. Dorn LD, Burgess ES, Dubbert B, Simpson SE, Friedman T, Kling M, et al. Psychopathology in patients with endogenous Cushing's syndrome: ‘atypical' or melancholic features. Clin Endocrinol. (1995) 43:433–42. doi: 10.1111/j.1365-2265.1995.tb02614.x
10. Anil Kumar BN, Grover S. Cushing's syndrome masquerading as treatment resistant depression indian. J Psychol Med. (2016) 38:246–8. doi: 10.4103/0253-7176.183095
11. Loosen PT, Chambliss B, DeBold CR, Shelton R, Orth D. Psychiatric phenomenology in Cushing's disease. Pharmacopsychiatry. (1992) 25:192–8. doi: 10.1055/s-2007-1014405
12. Sonino N, Fava GA, Belluardo P, Girelli ME, Boscaro, M. Course of depression in Cushing's syndrome: response to treatment and comparison with Graves' disease. Horm Res. (1993) 39:202–6. doi: 10.1159/000182736
13. Starkman MN, Schteingart DE, Schork M. A depressed mood and other psychiatric manifestations of Cushing's syndrome: relationship to hormone levels. Psychosom Med. (1981) 43:3–18. doi: 10.1097/00006842-198102000-00002
14. Al-Harbi SD, Mashi AH, Al Johani N. A case of Cushing's disease presenting with isolated suicidal attempt clin med insights. Case Rep. (2021) 14:11795476211027668. doi: 10.1177/11795476211027668
15. Fujii Y, Mizoguchi Y, Masuoka J, Matsuda Y, Abe T, Anzai K, et al. Cushing's syndrome and psychosis: a case report and literature review. prim care companion. CNS Disord. (2018) 20:279. doi: 10.4088/PCC.18br02279
16. Starkman MN, Giordani B, Berent S, Schork MA, Schteingart D. Elevated cortisol levels in Cushing's disease are associated with cognitive decrements. Psychosom Med. (2001) 63:985–93. doi: 10.1097/00006842-200111000-00018
Keywords: Cushing's syndrome (CS), treatment-resistant depression, acute psychosis, adrenal adenoma (AA), adolescent girl
Citation: Yin X, Chen S, Ju X and Hu X (2023) Case report: Treatment-resistant depression with acute psychosis in an adolescent girl with Cushing's syndrome. Front. Psychiatry 14:1170890. doi: 10.3389/fpsyt.2023.1170890
From https://www.frontiersin.org/articles/10.3389/fpsyt.2023.1170890/full
-
1
-
-
Abstract
The most common cause of Cushing syndrome (CS) is exposure to exogenous glucocorticoids. There is an increasing incidence of adulterated over-the-counter (OTC) supplements containing steroids. We present a case of Artri King (AK)-induced CS in a 40-year-old woman who presented with an intertrochanteric fracture of her right femur. Laboratory testing revealed suppressed cortisol and adrenocorticotropic hormone, which was consistent with suppression of the hypothalamic-pituitary-adrenal (HPA) axis. Following the cessation of the AK supplement, the patient’s HPA axis recovered, and the clinical manifestations of CS improved. This case emphasizes the need for better regulation of OTC supplements and the need for cautious use.
Introduction
Cushing syndrome (CS) is a condition that occurs because of high blood levels of glucocorticoids (GCs). These patients can present with a variety of systemic signs and symptoms, including truncal obesity, easy bruising of the skin, violaceous abdominal striae, resistant hypertension, dysglycemia, as well as osteoporosis. CS can occur because of adrenal etiologies such as adrenal adenoma, adrenal cancer, or adrenal hyperplasia or from an adrenocorticotropic hormone (ACTH)-producing pituitary adenoma or ectopic tumor. However, the most common cause of CS is the exogenous administration of GCs [1]. While exogenous GCs are often medically prescribed for the treatment of inflammatory conditions, some patients may be accidentally exposed to exogenous GCs from over-the-counter (OTC) supplements. We present a case of a young woman who developed exogenous CS and suffered a hip fracture as a result of taking an OTC supplement, Artri King (AK), adulterated with GCs.
Case Presentation
A 40-year-old obese woman presented to the hospital following a fall at home. She reported a snapping noise and sudden right hip pain while trying to stand up, and subsequently fell to the floor. She had noted right-sided hip pain for several days preceding her fall. She was evaluated in the emergency department where computed tomography (CT) imaging of the right lower extremity showed an intertrochanteric fracture of the right femur (Figure 1). The patient underwent open reduction and internal fixation of her right femur. The patient reported an unexplained weight gain of approximately 40 lbs in the preceding five months with a peak weight of 223 lbs (101 kg) and a body mass index (BMI) of 37 kg/m2. The patient denied taking any medications or supplements at the time of hospitalization. The endocrinology team was consulted to evaluate for causes of secondary osteoporosis in this young woman.
")
Figure 1: A CT scan showing the right intertrochanteric fracture of the right femur (yellow arrows)
Diagnostic assessment
Her vital signs showed a blood pressure of 142/96 mmHg, heart rate of 68 beats per minute, temperature of 98.1°F (36.7°C), and 98% oxygenation on room air. Physical examination did not reveal abdominal striae or buffalo hump. She did have supraclavicular fat deposition and central obesity. No proximal muscle weakness was present.
Laboratory tests were pertinent for decreased 25-hydroxy vitamin D, increased parathyroid hormone (PTH), and normal calcium (Table 1). These findings were consistent with secondary hyperparathyroidism due to vitamin D deficiency. Dual-energy X-ray absorptiometry (DEXA) scan revealed osteoporosis (Figures 2, 3 and Tables 2, 3). Further testing showed normal thyroid-stimulating hormone (TSH), estradiol, follicle-stimulating hormone (FSH), and luteinizing hormone (LH), thus ruling out hyperthyroidism and primary ovarian insufficiency as possible causes of reduced bone mineral density (Table 1). Random cortisol was checked as hypercortisolism was suspected but it was found to be decreased along with decreased ACTH as well (Table 4). A cosyntropin stimulation test was performed, which showed decreased baseline cortisol with inappropriately decreased cortisol levels at 30 minutes and 60 minutes (Table 5). Given the discordance between the patient’s presentation and the lab results, assay interference was suspected, and further evaluation of the adrenal function was performed. Repeat labs using liquid chromatography-mass spectrometry (LCMS) assay again confirmed persistently low cortisol (Table 4). A 24-hour free urine cortisol was too low to quantify per assay despite the adequate volume. Further evaluation showed overall low adrenal steroids, including deoxycorticosterone, 17-hydroxyprogesterone, androstenedione, 11-deoxycortisol, pregnenolone, dehydroepiandrosterone sulfate, corticosterone, and progesterone.
Lab test Patient's value Reference range 25-hydroxy vitamin D 12.8 ng/ml 30-100 ng/ml Parathyroid hormone (PTH) 86.2 pg/ml 10-66 pg/ml Serum calcium 9.5 ng/dl 8.8-10.5 mg/dl Thyroid-stimulating hormone (TSH) 2.49 mIU/L 0.36-3.74 mIU/L Estradiol 57.1 pg/ml 19.8-144.2 pg/ml Follicle-stimulating hormone (FSH) 5.4 mIU/ml 2.5-10.4 mIU/ml Luteinizing hormone (LH) 6 mIU/ml 1.9-12.5 mIU/ml Table 1: Patient's lab values on admission
-scan-of-the-femoral-neck-showing-osteopenia")
Figure 2: Dual-energy X-ray absorptiometry (DEXA) scan of the femoral neck showing osteopenia
-scan-of-the-lumbar-spine-showing-osteoporosis")
Figure 3: Dual-energy X-ray absorptiometry (DEXA) scan of the lumbar spine showing osteoporosis
Region Area (cm2) Bone mineral content (g) Bone mineral density (g/cm2) T-score Peak reference Z-score Age-matched Femoral neck 4.76 3.53 0.742 -1.0 87 -0.7 91 Total 33.39 26.14 0.783 -1.3 83 -1.1 85 Table 2: Summary of dual-energy X-ray absorptiometry (DEXA) scan results of the femoral neck
Region Area (cm2) Bone mineral content (g) Bone mineral density (g/cm2) T-score Peak reference Z-score Age-matched L1 10.79 7.56 0.701 -2.6 71 -2.4 73 L2 11.79 9.06 0.768 -2.4 75 -2.1 77 L3 12.70 9.98 0.786 -2.7 73 -2.4 75 L4 15.57 11.42 0.733 -3.0 69 -2.7 71 Total 50.86 38.03 0.748 -2.7 71 -2.5 73 Table 3: Summary of dual-energy X-ray absorptiometry (DEXA) scan results of the lumbar spine
Lab test Patient's values while on Artri King Patient's values four weeks off of Artri King Reference range Random cortisol (routine assay) <0.64 μg/dL 7.3 μg/dL 5-25 μg/dL Adrenocorticotropic hormone (ACTH) 1.5 pg/ml 12 pg/ml 7.2-63.3 pg/ml Random cortisol (using liquid chromatography-mass spectrometry (LCMS) assay) 0.526 μg/dL N/A 5-25 μg/dL Table 4: Patient's cortisol and adrenocorticotropic hormone levels before and after stopping Artri King
Cosyntropin stimulation test Patient value Reference range Baseline cortisol 1.64 μg/dL 5-25 μg/dL Cortisol after 30 minutes 1.33 μg/dL >18 μg/dL Cortisol after 60 minutes 6.48 μg/dL >18 μg/dL Table 5: Results of cosyntropin test while on Artri King
Treatment
She was started on teriparatide as well as vitamin D and calcium supplementation for the treatment of osteoporosis. Based on the aforementioned testing and the apparent symptoms of hypercortisolism, the patient was questioned again about the potential intake of steroids. She then recalled that she had been taking AK, an OTC supplement promoted for joint pain and arthritis. She reported that she had been taking two tablets of the supplement three times a day intermittently for the past three years. The patient neglected to bring it to the medical team’s attention before because she was under the impression that it was a multivitamin and did not have implications on her diagnosis. She was asked to stop the supplement and was educated about potential adrenal insufficiency symptoms and GC withdrawal.
Outcome and follow up
Repeat labs after four weeks off AK showed improved cortisol and ACTH levels indicating recovery of her hypothalamic-pituitary-adrenal (HPA) axis (Table 4). She lost 25 lbs in this time span with lifestyle modification. She continues teriparatide for osteoporosis, and monitoring of her bone mineral density is planned.
Discussion
This patient initially presented with a pathological fracture of her right femoral head. Given her young age, causes of secondary osteoporosis, including CS, were explored. The prevalence of osteoporosis in CS patients is 50% [2]. The effects of GC on bone health have been well studied. The major mechanism by which GC affects bone mineral density is by impairment of bone formation. GCs increase osteoblast and osteocyte apoptosis and decrease osteoblast function through their catabolic effects, which result in a dramatic decrease in bone formation rate. A prolonged lifespan of osteoclasts is observed with GC. A decrease in bone formation markers such as P1NP and osteocalcin has been observed in patients treated with GC [3]. Long-term GC use is associated with increased risk for fractures with a reported global prevalence of fractures of 30-50%. The risk for vertebral fractures is even higher, particularly in the thoracic and lumbar vertebrae. Interestingly, the risk for fracture with GC use peaks early in the course of treatment, often as early as three months into treatment, and declines rapidly after GC discontinuation [4]. An increased fracture risk has been described even with relatively low doses of GC (2.5-7.5 mg of prednisone or other equivalently dosed GC) and even with short-term use of under 30 days [5].
Our patient’s initial labs confirmed adrenal suppression despite our initial suspicion of CS, given her ongoing weight gain, central obesity, and osteoporosis. However, no obvious source of exogenous GC was identified. In most cases, the source of exogenous GC is easily identified through medication reconciliation; however, in our case, the patient was inadvertently exposed to steroids from an unregulated supplement, AK. The supplement’s ingredients were listed as glucosamine, chondroitin, collagen, vitamin C, curcumin, methylsulfonylmethane, nettle, and omega-3 fatty acids, with no mention of any steroid components. In a letter to the editor of the Internal Medicine magazine, several doctors published their concerns about a recent increase in CS cases associated with the use of AK and other similarly unregulated products [6]. Based on our literature search, three similar cases were published [7,8]. The reported cases developed CS after taking Artri King for several months, but none of them presented with a fracture.
A warning by the U.S. Food & Drug Administration (FDA) was issued on April 20, 2022, indicating that FDA laboratory testing of this supplement confirmed the presence of undeclared drug ingredients, including dexamethasone, methocarbamol, and diclofenac. The FDA, however, was unable to confirm the exact amount of dexamethasone that these supplements contained [9]. Adverse events, including liver toxicity and death, were reported by the FDA.
One study revealed that between 2007 and 2016, the FDA had issued more than 700 warnings about the sale of dietary supplements that contained unlisted and potentially dangerous ingredients. The majority of these supplements included those marketed for sexual enhancement, weight loss, or muscle building [10]. This case highlights the risks of undisclosed ingredients in OTC supplements.
Conclusions
In conclusion, we recommend that a thorough reconciliation of medication and supplements be obtained for all patients with CS. Supplements should be stopped and HPA axis testing should be repeated in patients with suspected exogenous GC exposure, even if steroids are not declared in the ingredients. It is also important to monitor such patients for adrenal insufficiency due to GC withdrawal and consider GC tapering if necessary. Our patient showed improvement in cortisol levels with no overt symptoms of adrenal insufficiency without the need for GC therapy. This case demonstrates the first case of AK-induced CS resulting in a pathological fracture. Given the increased use and availability of OTC supplements, this case highlights on the importance of detailed history-taking and the role of supplements in causing CS. This case also stresses the need for further education and counseling of our patients as well as tighter control on the manufacturing and sale of these supplements.
References
- Lacroix A, Feelders RA, Stratakis CA, Nieman LK: Cushing's syndrome. Lancet. 2015, 386:913-27. 10.1016/S0140-6736(14)61375-1
- Mancini T, Doga M, Mazziotti G, Giustina A: Cushing's syndrome and bone. Pituitary. 2004, 7:249-52. 10.1007/s11102-005-1051-2
- Briot K, Roux 😄 Glucocorticoid-induced osteoporosis. RMD Open. 2015, 1:e000014. 10.1136/rmdopen-2014-000014
- Canalis E, Mazziotti G, Giustina A, Bilezikian JP: Glucocorticoid-induced osteoporosis: pathophysiology and therapy. Osteoporos Int. 2007, 18:1319-28. 10.1007/s00198-007-0394-0
- Waljee AK, Rogers MA, Lin P, et al.: Short term use of oral corticosteroids and related harms among adults in the United States: population based cohort study. BMJ. 2017, 357:j1415. 10.1136/bmj.j1415
- Del Carpio-Orantes L, Quintín Barrat-Hernández A, Salas-González A: Iatrogenic Cushing syndrome due to fallacious herbal supplements. The case of Ortiga Ajo Rey and Artri King. Med Int Mex. 2021, 37:599-602.
- Patel R, Sherf S, Lai NB, Yu R: Exogenous Cushing syndrome caused by a "Herbal" supplement. AACE Clin Case Rep. 2022, 8:239-42. 10.1016/j.aace.2022.08.001
- Mikhail N, Kurator K, Martey E, Gaitonde A, Cabrera C, Balingit P: Iatrogenic Cushing’s syndrome caused by adulteration of a health product with dexamethasone. JSM Clin Case Rep. 2022, 3:
- U.S. Food and Drug Administration. Public notification: Artri King contains hidden drug ingredients. (2022). Accessed: February 25, 2023: https://www.fda.gov/drugs/medication-health-fraud/public-notification-artri-king-contains-hidden-drug-ingredients.
- Tucker J, Fischer T, Upjohn L, Mazzera D, Kumar M: Unapproved pharmaceutical ingredients included in dietary supplements associated with US Food and Drug Administration warnings. JAMA Netw Open. 2018, 1:e183337. 10.1001/jamanetworkopen.2018.3337
-
1
-
Nearly one-third of women with endogenous Cushing’s syndrome and normal bone mineral density have a low trabecular bone score, according to study data.
“A large proportion of patients had degraded microarchitecture despite normal BMD,” Hiya Boro, DM, MD, MBBS, consultant in endocrinology, diabetes and metabolism at Aadhar Health Institute in India, and colleagues wrote. “The risk of fracture may be underestimated if BMD alone is measured. Hence, trabecular bone score should be added as a routine complementary tool in the assessment of bone health in patients with Cushing’s syndrome.”
 Data were derived from Boro H, et al. Clin Endocrinol. 2023;doi:10.1111/cen.14944.
Data were derived from Boro H, et al. Clin Endocrinol. 2023;doi:10.1111/cen.14944.
Researchers conducted a cross-sectional study at a single center in India from March 2018 to August 2019. The study included 40 women with overt endogenous Cushing’s syndrome and 40 healthy sex-matched controls. Seum and salivary cortisol and plasma adrenocorticotropic hormone (ACTH) were measured. Participants were considered ACTH independent if they had a level of less than 2.2 pmol/L. Areal BMD was measured at the lumbar spine, femoral neck, total hip and distal one-third of the nondominant distal radius. Low BMD for age was defined as a z score of less than –2. Trabecular bone score was measured at the lumbar spine. Fully degraded microarchitecture was defined as a trabecular bone score of 1.2 or lower and partial degradation was a trabecular bone score of 1.21 to 1.34.
Of the 40 women with Cushing’s syndrome, 32 were ACTH-dependent and the other eight were ACTH independent. Of the independent group, seven had adrenal adenoma and one had adrenocortical carcinoma.
Women with Cushing’s syndrome had lower BMD at the lumbar spine (0.812 g/cm2 vs. 0.97 g/cm2; P < .001), femoral neck (0.651 g/cm2 vs. 0.773 g/cm2; P < .001) and total hip (0.799 g/cm2 vs. 0.9 g/cm2; P < .001) than the control group.
“No significant difference was noted in the distal radius BMD,” the researchers wrote. “This may be explained by the fact that cortisol excess predominantly affects trabecular rather than cortical bone.”
Absolute trabecular bone score was lower in the Cushing’s syndrome group compared with controls (1.2 vs. 1.361; P < .001). Based on trabecular bone score, 42.5% of women with Cushing’s syndrome had fully degraded bone microarchitecture, 45% had partially degraded microarchitecture and 12.5% had normal microarchitecture.
“In our study, 32.5% of patients had normal BMD with low trabecular bone score, thus highlighting the fact that patients may have normal BMD despite degraded microarchitecture,” the researchers wrote. “As such, assessment of BMD alone may underestimate the risk of fractures in patients with Cushing’s syndrome.”
-
Abstract
Context
Cushing’s disease (CD) is rare condition burdened by several systemic complications correlated to higher mortality rates. The primary goal of clinicians is to achieve remission, but it is unclear if treatment can also increase life expectancy.
Aim
To assess the prevalence of cortisol-related complications and mortality in a large cohort of CD patients attending a single referral centre.
Materials and methods
The clinical charts of CD patients attending a referral hospital between 2001 and 2021 were reviewed.
Results
126 CD patients (median age at diagnosis 39 years) were included. At the last examination, 78/126 (61.9%) of the patients were in remission regardless of previous treatment strategies. Patients in remission showed a significant improvement in all the cardiovascular (CV) comorbidities (p < 0.05). The CV events were more frequent in older patients (p = 0.003), smokers and persistent CD groups (p < 0.05). Most of the thromboembolic (TE) and infective events occurred during active stages of the disease. The CV events were the most frequent cause of death. The standardized mortality ratio (SMR) resulted increased in persistent cases at the last follow-up (SMR 4.99, 95%CI [2.15; 9.83], p < 0.001) whilst it was not higher in those in remission (SMR 1.66, 95%CI [0.34; 4.85], p = 0.543) regardless of the timing or number of treatments carried out. A younger age at diagnosis (p = 0.005), a microadenoma (p = 0.002), and remission status at the last follow-up (p = 0.027) all increased survival. Furthermore, an elevated number of comorbidities, in particular arterial hypertension, increased mortality rates.
Conclusions
Patients with active CD presented a poor survival outcome. Remission restored the patients’ life expectancy regardless of the timing or the types of treatments used to achieve it. Persistent CD-related comorbidities remained major risk factors.
Introduction
Cushing’s disease (CD) is the most common cause of endogenous glucocorticoid excess due to uncontrolled adrenocorticotropic hormone (ACTH) secretion from a pituitary adenoma, for the most part a microadenoma [1]. A rare condition with an estimated incidence of 0.6—2.6 cases per million per year, it is burdened by high morbidity and mortality, for the most part linked to cardiovascular (CV) events. This is particularly true for active CD which is characterized by hypertension, diabetes mellitus, obesity and dyslipidaemia. The severity of the clinical picture seems to depend more on the duration of the disease rather than on the degree of cortisol elevation, although other confounding factors may affect the clinical phenotype [2]. Prompt diagnosis and resolution of hypercortisolemia are paramount to revert cortisol-related comorbidities and to improve life expectancy. Although new individualized medical treatment options for CD continue to evolve, transsphenoidal surgery (TSS) remains the first line treatment for potentially operable patients as it is the only treatment that seems to provide a rapid, long-lasting remission. Persistent and recurrent cases are nevertheless major concerns, since up to 50% of cases might require other treatment modalities to achieve disease control and those patients are once again exposed to cortisol excess that can negatively impact their survival [3]. An increased mortality has been noted in patients with active CD, while patients in remission show a markedly lower one. It is still unclear if mortality in these patients is higher than that in the general population. Some studies report a normal life expectancy [4,5,6,7,8] while others describe a persistently higher mortality [9,10,11]. One study reported finding a higher mortality as long as 10 years after remission, and only patients cured by a single TSS showed a normal life expectancy [12].
In view of these considerations, this study was designed to assess the prevalence of cortisol-related comorbidities/complications and mortality in a large group of CD patients attending a tertiary referral centre over the past 20 years. Other study aims were to evaluate the predictors of long-term outcomes and the impact of different treatments on life expectancy in CD patients.
Materials and Methods
One hundred twenty-six CD patients diagnosed between December 2001 and December 2021 were eligible for this monocentric, retrospective, observational study. Hypercortisolism was suspected on the basis of the patient’s clinical features and it was confirmed by appropriate hormonal testing [low dose dexamethasone suppression test (LDDST), 24-h urinary free cortisol (UFC) and late-night salivary cortisol (LNSC)] after excluding the possibility of exogenous glucocorticoid intake from any route [13]. UFC and LNSC were assessed at least in two different samples as recommended [14, 15].
The diagnosis of ACTH-dependent syndrome was confirmed on the strength of detectable ACTH levels (> 10 ng/L) and appropriate responses to a high dose dexamethasone suppression test (HDDST), corticotrophin releasing hormone (CRH) and/or desmopressin (DDAVP) tests [16]. All the patients underwent a pituitary magnetic resonance imaging (MRI); they also underwent bilateral inferior petrosal sinus sampling (BIPSS) when the results of hormonal tests were ambiguous. The pituitary origin of ACTH secretion was confirmed by biochemical remission after TSS, histology and/or post-operative hypoadrenalism.
The results of clinical, biochemical and radiological tests as well as the treatments performed to control cortisol secretion (surgery, radiotherapy and/or medical therapy), any comorbidities (i.e., arterial hypertension, impaired glucose homeostasis, dyslipidaemia, overweight), any hormone deficiencies, any complications (i.e., CD-related events such as infective, CV and thromboembolic events) and any deaths recorded in the medical charts were collected.
The disease severity at baseline was defined on the basis of the patient’s UFC values as mild (up to two-fold the upper limit of normal – ULN), moderate (between 2 and 5 times the ULN) or severe (over five-fold the ULN).
Patient’s classification on the basis of disease activity are indicated in Supplementary material and methods sections.
The presence of hypertension, glucose metabolism impairment, obesity, dyslipidaemia and hypopituitarism were defined as by specific Guidelines, Supplementary [19,20,21,22,23,24].
The current study was designed in accordance with the principles of the Declaration of Helsinki and approved by the Ethical Committee of the province of Padova (protocol code 236n/AO/22, date of approval 29 April 2022).
The types of CD complications characterizing the patient were classified into three categories: CV, thromboembolic (TE), or infective (IN) events. Depending on the timing of its presentation, an event was classified as occurring: “prior” to diagnosis, “during” active CD or “after” CD remission. Events requiring hospitalization or iv antibiotic administration were registered as IN events. The causes of death were classified under the following headings: CV, infections, cancer, psychiatric complications leading to suicide, TE events or other (the last when none of the previous causes was applicable).
Statistical analysis
Categorical variables were reported as counts or percentages, and quantitative variables as median and interquartile ranges [IQR]. The comparisons between groups were performed with a Mann–Whitney sum rank test for independent quantitative variables; a Wilcoxon signed-rank test was run for dependent quantitative variables. As far as categorical variables were concerned, the McNemar test or a chi-square test were used for paired and unpaired data, respectively.
A Cox regression analysis was performed to evaluate possible predictors for events and mortality based on the assumption of constant hazards over time. As time-dependent variables (e.g., achieving remission) did not meet this assumption, their survival analysis was performed using Kaplan–Meier analysis. Regarding complications, as there is usually a delay in CD diagnosis [25], Kaplan Meier curves for event free probability were calculated beginning 24 months prior to the diagnosis in order to include “prior” events possibly related to cortisol excess in our analysis. Vice versa, survival analysis for mortality was calculated beginning with the CD diagnosis date. Standardized mortality ratio (SMR) was calculated based on indirect age standardization in order to compare the observed deaths in our CD population with the expected number of deaths in the general population [26, 27]. A Fisher exact test was carried out to assess significant differences with respect to the general population and calculating the 95% confidence interval (95% CI) for SMR.
The threshold for statistical significance was set at p-value < 0.05. Statistical analyses were performed with R: R-4.2.0 for Windows 10 (32/64 bit) released in April 2022 and R studio desktop version 4.2.0 (2022-04-22) for Windows 10 64 bit (R Foundation for Statistical Computing, Vienna, Austria, URL https://www.R-project.org/). An open-source calculator was also used to perform the Fisher exact test (http://www.openepi.com).
Results
Baseline
The data of 167 CD patients attending the Centre between December 2001 and December 2021 were collected. The information regarding 41 patients were not included in the analysis because of insufficient follow-up data (i.e. patients referred for second opinion or for diagnostic workup or those with follow-up < 1 year from first line treatment). The remaining 126 patients presented a median age at diagnosis of 39 [31–50 years]; the female: male ratio was 3:1. The median follow-up was 130.5 months [72.5–201.5]. The patients’ clinical features at the time of diagnosis are outlined in Table 1.
Table 1 The patients’ clinical features at the time of diagnosis The median UFC levels were 3.2 times the ULN [2–5.6]. Almost half of the cohort presented moderate cortisol excess (45/98, 45.9%), with lower proportions of the patients presenting mild (26/98, 26.5%) and severe disease (22/98, 27.6%).
Most of the patients (91/113, 80.5%) had a microadenoma, including 29/91(31.9%) with negative imaging. The remaining 22 patients (19.5%) had a macroadenoma.
Treatments
Most of the patients underwent TSS as the first line treatment (113/126), only one patient underwent craniotomy. Eight patients received primary medical treatment, three received first-line radiotherapy and one underwent BA soon after diagnosis. Overall, 115 patients underwent pituitary surgery (one patient with a previous unsuccessful pituitary irradiation) and the remission rate was 60.9%. Relapses were observed in 46.7% of the cases after a median time of 56 [29–83] months. The second surgery proved less successful with respect to the first one; the remission rate was 43.2% (16/37); of these, 25% developed recurrence during the follow-up period. The median time to relapse was 66.5 [36–120] months. Only two patients underwent a third surgery; in both cases it was not curative (Supplementary Fig. 1) [27]. A 4th and a 5th TSS were performed in one of these for debulking purposes due to an aggressive pituitary lesion. Surgical remission was not affected by pre-treatment with cortisol-lowering medications neither before the first (p = 1.0) nor the second TSS (p = 0.88). Moreover, hormone control did not improve the surgical outcomes, although a tendency towards a higher remission rate was observed in those patients who showed good disease control before undergoing the second surgery (Supplementary Fig. 2) [27].
Overall, 34 patients received radiotherapy, either the conventional (18.5%) or the stereotactic type (81.5%). Remission was noted in 36.7% (11/30) of the patients with at least a 12-month post-radiotherapy follow-up. As expected, the longer the follow-up, the higher the remission rate; it was 41.67% (10/24) and 46.7% (7/15) at 5 and 10 years, respectively.
Thirteen patients underwent BA and achieved complete remission. Excluding the patients with less than 12 months of follow-up, 4 out of 11 (36.4%) of the patients developed CTP-BADX/NS over a mean follow-up period of 110 [106 -329] months. Three patients out of the 11 were previously irradiated at pituitary level to control cortisol secretion. Four CD patients underwent unilateral adrenalectomy due to a dominant adrenal lesion consistent with chronic ACTH stimulation. Two (50%), harbouring unilateral adenomas larger than 5 cm, achieved remission after surgery; both cases were previously irradiated at the pituitary level.
All but one of the 48 patients with persistent hypercortisolism at the last follow-up were on cortisol lowering medications. The untreated patient had a residual mild cortisol excess after TSS and medical therapy was discontinued because of multiple drug intolerance. At the last follow-up 28 patients were receiving monotherapy, and 19 were receiving combination treatment; 25 patients were receiving steroidogenesis inhibitors, 9 pituitary-target drugs and 13 a combination of the two compounds (Supplementary Table 1) [27]. Most of our patients achieved UFC normalization (complete control in 67.4%, partial control in 22.7%, uncontrolled in 10.9%). Data pertaining to a single patient with renal function impairment who presented falsely low UFC were not included in this analysis. When available, LNSC was restored in 14/41 cases (34.2%). No differences in the patients’ outcomes linked to the type of treatment prescribed (monotherapy vs combination treatment) or its target (adrenal vs pituitary) were found (data not shown).
We also evaluated the extent of cortisol excess throughout the active phase of CD both for the patients presenting persistence at the last available follow-up (n = 48) and for those in remission after multiple therapies (i.e., late remission) (n = 33). As described in the material and methods section, disease activity for each year of active disease was defined on the basis of patients’ UFC levels. A minimum of three UFC measurements were registered every year and the median value was calculated. When data were missing, the patients were considered uncontrolled during that period. The results are reported in Supplementary Table 2 [27]; both the persistence and late remission groups showed UFC levels < 2xULN over more than 50% of the time span evaluated (58.8% and 73.6%, respectively). There was a progressive increase in the proportion of controlled patients over the observation period (Fig. 1).
Fig. 1 
Percentage of patients controlled during active CD
Comorbidities
The principal CD features at baseline and at the last follow-up examination were evaluated, (Supplementary Table 2). At time of diagnosis, no differences were observed as regards comorbidities between patients who achieved remission and those with persistent disease at baseline, (Supplementary Table 3). The patients in remission at the last examination showed a significant improvement in all the parameters considered; those with persistent CD did not (Table 2).
Table 2 A comparison of Cushing’s disease features at baseline and at the last follow-up examination As far as hormone deficiencies were concerned, 42/126 (33.3%) of the patients developed at least one deficit due to previous treatments (Supplementary table 4) [27], including hypocortisolism due to BA. Neither the second surgery nor radiotherapy led to an increase in hypopituitarism (Supplementary Fig. 3) [27].
Complications and mortality
As far as CD complications were concerned, 18.3% of the patients had a TE event, 17.5% presented an IN event and 7.1% presented a CV one. Most of the events occurred during an active phase of CD (Table 3). Other concomitant thrombotic risk factors were present in 10/19 (52.6%) of the patients experiencing TE events. TE events were related to surgery (pituitary, adrenal or others) in 5 cases, to post-traumatic fractures in 2, to prolonged immobilization in 2, and to a symptomatic SARS CoV2 infection in one case. IN events affected the respiratory system in 9 cases, the gastro-intestinal tract in 5 cases, the soft tissues in three cases, the central nervous system in 2 cases, the musculoskeletal system in 2 cases and the genitourinary tract in one case.
Table 3 Thromboembolic, infective, and cardiovascular events and their timing (see materials and methods) Overall, 11 deaths were recorded during the follow-up period (130.5 [72.5–201.5] months). The causes of death were classified as: cardiovascular events (n = 4), infections (n = 2), cancer (n = 2), suicide (n = 1), thromboembolic events (n = 0), others (n = 2; a cerebral haemorrhage in one case and an unknown cause in the other).
Cox regression was performed to evaluate the predictors of events (CV, IN, TE) and mortality (Fig. 2). The older patients presented an increased risk of mortality (HR 9.41, 95%CI [1.97; 44.90], p = 0.005), of CV events (HR 4.84, 95%CI [1.13; 20.75], p = 0.003) and of TE events (HR 2.41, 95%CI [1.02; 5.65], p = 0.04). Similarly, the presence of a macroadenoma at the time of the first MRI was associated with reduced survival (HR 9.29, 95%CI [2.30; 37.53], p = 0.002). Smoking was correlated to CV events (HR 5.33, 95%CI [1.33; 21.37], p = 0.02). Hypercortisolism severity at baseline did not affect the risk of complications or survival. No gender related differences were observed, although a tendency toward more CV events was noted in the males (p = 0.08).
Fig. 2 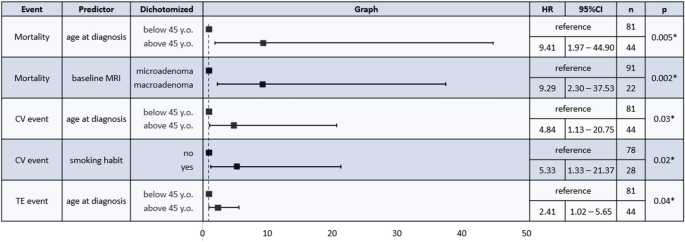
Cox regression analysis for predictors of mortality and cardiovascular, infective or thromboembolic events; only significant results are shown. HR: Hazard ratio; CI: confidence interval; n: number, CV: cardiovascular; TE: thromboembolic. *p < 0.05
Kaplan Meier curves were plotted for complications (CV, IN and TE) and mortality in order to assess time-dependent variables (i.e., the number of comorbidities and the disease status at the last follow-up, the timing of remission and the disease activity in the patients with persistent CD at the last follow-up). We found that persistent disease and multiple comorbidities (at least 3) at the last follow-up were associated with increased CV events (p = 0.044 and p = 0.013, respectively) and mortality (p = 0.027 and p = 0.0057, respectively) (Fig. 3). The timing of remission did not influence the mortality or the risk of complications (data not shown). With regard to the patients with persistence, those presenting total/partial control for more than half of the follow-up period considered tended to have fewer CV and IN events (p = 0.078 and p = 0.074, respectively) (Fig. 3). Similarly, among patients with persistent cortisol excess the impaired circadian rhythm of secretion was associate to TE events and a trend to higher mortality (Supplementary Fig. 4). Sub-analysis of each comorbidity revealed that hypertension played a pivotal role during the follow-up period for CV complications (p = 0.011) and mortality (p = 0.0039). Similarly, dyslipidaemia was related to CV events (p = 0.046) and prediabetes/diabetes were associated to TE events (p = 0.035). A tendency toward increased mortality in the patients with impaired glucose homeostasis at the last follow-up was also noted (p = 0.052) (Data not shown).
Fig. 3 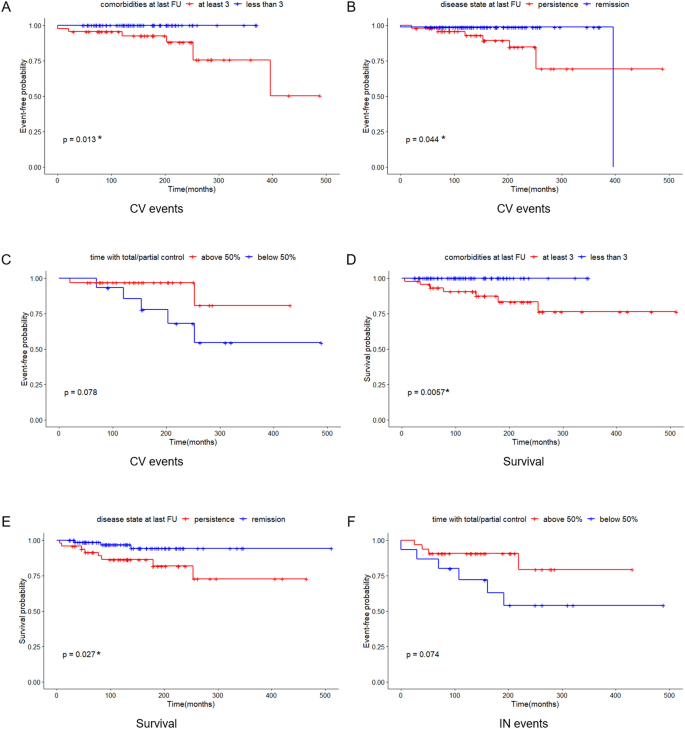
Kaplan Meier curves for cardiovascular events based on: A) comorbidities at the last follow-up examination;
disease status at the last follow-up examination; C) control during active disease for patients presenting persistence at the last follow-up. Kaplan Meier curves for survival plotting: D) comorbidities at the last follow-up examination; E) disease status at the last follow-up examination. Kaplan Meier curves for infective events based on: F) hormone control during active disease of patients presenting persistence at the last follow-up examination. FU: follow-up; CV: cardiovascular; IN: infective. *p < 0.05
The entire CD cohort presented an increased mortality, with a SMR of 3.22 (95%CI [1.70; 5.60], p = 0.002). Mortality was significantly higher in the patients with persistent disease (SMR 4.99, 95%CI [2.15; 9.83], p < 0.001), but it was similar to that of the general population in the patients in remission (SMR 1.66, 95%CI [0.34; 4.85], p = 0.543). The finding was independent of the timing or the modality used to achieve cortisol control; for the early remission group the SMR was 2.15 (95%CI [0.36; 7.11], p = 0.477) and for the late remission group it was 1.14 (95%CI [< 0.01; 5.62], p = 1.0). The length of remission period was 82 [38–139] for the early remission group vs 85 [21–136] for the late remission one.
Discussion
Study findings have confirmed that CD patients have a higher mortality and, as previously observed, the most common cause of death in these patients was, first of all, CV events and, secondly, infections [9]. Although there were no fatal TE events in our cohort, that type of complication was the most frequent one. As expected, the patients with persistent CD presented significantly increased mortality with respect to the general population. At the last follow-up examination the CD patients in remission had a mortality rate that was comparable to that of the general population regardless of the number of treatments needed to achieve remission. The finding is in contrast with the results of a multicentre study examining patients with more than 10 years of remission that reported finding a normal life expectancy only in the patients who achieved an early remission following a single TSS [12]. The better life expectancy in our series may be explained by an extensive use of cortisol-lowering medications in our centre during active phases of CD. There was moreover at least a partial control in the late remission group during over 70% of the years assessed; this might have had a positive effect on the overall survival rate (data not shown). Furthermore, our study considered relatively recent years when significant improvement in timely diagnosis and available medical therapies have been made [9]. Lastly, being monocentric, our study showed a homogenous management of comorbidities that by contrast, is in highly unlikely in a retrospective international study. Since cardiovascular and metabolic risk factors related to cortisol-excess are major determinant of mortality in CD, the latter point is of the outmost importance.
Survival was positively influenced in our cohort by a younger age at diagnosis, the presence of a microadenoma at baseline [9] and a remission status at the last follow-up examination. As expected, an elevated number of comorbidities increased mortality, and as has been previously reported, arterial hypertension, in particular, reduced survival [28]. A tendency toward increased mortality was also noted in connection to impaired glucose homeostasis, but data on this topic are still controversial [8, 10, 12, 28, 29].
Cortisol excess atherosclerotic risk leading to CV events are closely liked. Beyond cortisol’s direct action on the tissues, this association is probably related to a clustering of several metabolic complications such as insulin resistance, arterial hypertension, dyslipidaemia and overweight commonly present in CD patients [30, 31]. Indeed, the patients presenting multiple comorbidities, especially arterial hypertension and dyslipidaemia, showed more CV complications. CV events were also more frequent in the patients with persistent hypercortisolism, and, as observed in general population in the elderly and in the smokers [32].
Older age at the time of diagnosis and dis-glycemia at the last follow-up examination were found to be related to TE events. It was instead impossible to identify predictors of infective complications. Although most TE and IN events occurred during active disease, remission did not significantly reduce these complications. The finding is in line with the data of a recent study focusing on a Swedish population reporting that CD patients present a higher risk of sepsis and thromboembolism even during long term remission [33]. Moreover, it is worthy of note that most of the TE events (52.6%) were accompanied by a concomitant risk factor such as recent surgery. These data highlight the importance of adequate prophylaxis in CD patients facing prothrombotic conditions such as those linked to a perioperative period [3, 34]. Disease severity at the baseline did not affect the patients’ complications or survival; the finding is not entirely surprising as the degree of cortisol excess does not necessarily correlate with the severity of the clinical picture [2].
The patients who achieved remission in our cohort showed an overall improvement in all the cortisol-related comorbidities. Hypertension was the most prevalent complication at the time of diagnosis, while overweight, which persisted in approximately 50% of the cases after remission, became by far the most frequent comorbidity. Glucose homeostasis alterations were the least prevalent at the time of diagnosis, although an underestimation is probable, as only fasting glycaemia or glycosylated haemoglobin were evaluated in most cases and provocative testing for hypercortisolism was not carried out [35].
With regards to demographic features, for the most part our patients were diagnosed during their third/fourth decade of life and they were prevalently female, in line with previous reports [36]. Most cases were due to a pituitary microadenoma (80% of the cases in our patients), including non-visible lesions on the MRI.
As far as treatment was concerned, the remission rate after the first TSS was quite low with respect to what would be expected at a tertiary centre; the finding can be explained by the fact that many of the patients studied had been referred to our unit after undergoing unsuccessful pituitary surgery elsewhere. However, the assessment of surgical performance in various centres goes beyond the aim of the present study. As expected, a second TSS was less successful than the first one, but the rate of success found in our patients was in line with literature data [37]. Although the immediate remission rate after a second TSS was comparable to the long term outcome of radiotherapy, a quarter of the patients experienced a relapse just as they did after the first surgery [17]. Regarding the risk of developing hypopituitarism was concerned, no significant difference was found between the two approaches. These data have confirmed that both re-intervention and radiation treatment can be considered valid second-tier options, and a case by case approach should be adopted. Pre-operative medical treatment with cortisol-lowering medications did not improve the surgical outcomes, regardless of its effectiveness in controlling cortisol excess, in line with data by the European Registry on Cushing’s Syndrome (ERCUSYN) [38].
At the last follow-up examination, no differences in disease control were found when the treatment targets (pituitary vs adrenal) of the patients were compared. A higher control rate of hypercortisolism during active CD was found over time, possibly reflecting better drug dose titration and the widening landscape of available drugs with over two thirds of the patients presented completely controlled UFC at last examination. The fact that only one third of our patients achieved circadian rhythm restoration confirmed the previously reported difficulty in normalizing this parameter [39,40,41]. Interestingly, TE were more frequent when LNSC was uncontrolled and the same tendency was observed for survival, confirming the better outcome of patients with rhythm restoration [8]. Although only the last available value of LNSC was assessed, this finding might potentially turn the spotlight on the importance of LNSC normalization during medical treatment [42], but further studies are required to confirm these data.
In line with previous reports, more than one third of the patients who underwent BA developed CTP-BADX/NS [18]. Although BA seems to immediately control hypercortisolism, this benefit should be carefully weighed against the risk of permanent adrenal insufficiency and CTP-BADX/NS. The patients received minimal doses of glucocorticoid replacement treatments following BA to avoid both over- and under treatment that might negatively impact survival [43], and this might explain why BA was not associated to increased mortality as observed in other series [44]. Unilateral adrenalectomy was performed in selected cases when a large adrenal nodule, probably provoked by chronic ACTH stimulation [45], was found. Interestingly, two patients who had previously undergone radiation treatment of the pituitary achieved disease remission after this surgery. The “transition” from pituitary to adrenal hypercortisolism after long standing ACTH-stimulation on adrenal nodules in CD patients has already been described by other investigators, and it may explain our findings in the patients studied [46].
The study’s retrospective single-centre nature represents its primary limitation. Its other important limitation, the relatively low number of cases and deaths examined, is of course linked to the condition’s rarity. Being a monocentric study does, on the other hand, have its advantages as it ensures that the treatment strategies, comorbidities evaluation and management are homogeneous. Furthermore, data on comorbidities, disease activity, type of cortisol lowering medications and comorbidities are available for most of our cohort. Besides, a potential protective effect of tailored medical therapy to reduce cortisol levels seems to reduce some complications and, to a less extent, overall mortality, especially when circadian cortisol secretion is restored. Further studies are still required to confirmed these latter findings.
To conclude, active CD is characterized by increased morbidity and mortality, but disease remission seems to restore a normal life expectancy regardless of the timing and type of treatment used to achieve it. Thus, our aim as physicians is to pursue this goal by any means. Conversely, persistent cases seem to maintain an increase mortality, despite the use of effective cortisol lowering medications. Clearly persistent CD-related comorbidities require opportune monitoring and prompt management.
Data availability
Raw data are available from the corresponding author upon reasonable request.
References
-
Barbot M, Zilio M, Scaroni C (2020) Cushing’s syndrome: overview of clinical presentation, diagnostic tools and complications. Best Pract Res ClinEndocrinolMetab 34(2):101380. https://doi.org/10.1016/j.beem.2020.101380
-
Guarnotta V, Amato MC, Pivonello R et al (2017) The degree of urinary hypercortisolism is not correlated with the severity of cushing’s syndrome. Endocrine 55(2):564–572. https://doi.org/10.1007/s12020-016-0914-9
-
Fleseriu M, Auchus R, Bancos I et al (2021) Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol 9(12):847–875. https://doi.org/10.1016/S2213-8587(21)00235-7
-
Jones PS, Swearingen B (2022) Pituitary surgery in Cushing’s disease: first line treatment and role of reoperation. Pituitary 25(5):713–717. https://doi.org/10.1007/s11102-022-01254-8
-
Dekkers OM, Biermasz NR, Pereira AM et al (2007) Mortality in patients treated for Cushing’s disease is increased, compared with patients treated for nonfunctioning pituitary macroadenoma. J ClinEndocrinolMetab 92(3):976–981. https://doi.org/10.1210/jc.2006-2112
-
Hassan-Smith ZK, Sherlock M, Reulen RC et al (2012) Outcome of Cushing’s disease following transsphenoidal surgery in a single center over 20 years. J ClinEndocrinolMetab 97(4):1194–1201. https://doi.org/10.1210/jc.2011-2957
-
Yaneva M, Kalinov K, Zacharieva S (2013) Mortality in Cushing’s syndrome: data from 386 patients from a single tertiary referral center. Eur J Endocrinol. 169(5):621–627. https://doi.org/10.1530/EJE-13-0320
-
Roldán-Sarmiento P, Lam-Chung CE, Hinojosa-Amaya JM et al (2021) Diabetes, active disease, and afternoon serum cortisol levels predict cushing’s disease mortality: a cohort study. J ClinEndocrinolMetab 106(1):e103–e111. https://doi.org/10.1210/clinem/dgaa774
-
Limumpornpetch P, Morgan AW, Tiganescu A et al (2022) The effect of endogenous cushing syndrome on all-cause and cause-specific mortality. J ClinEndocrinolMetab 107(8):2377–2388. https://doi.org/10.1210/clinem/dgac265
-
Ragnarsson O, Olsson DS, Papakokkinou E et al (2019) Overall and disease-specific mortality in patients with cushing disease: a swedish nationwide study. J ClinEndocrinolMetab 104(6):2375–2384. https://doi.org/10.1210/jc.2018-02524
-
Bengtsson D, Ragnarsson O, Berinder K et al (2022) Increased mortality persists after treatment of cushing’s disease: a matched nationwide cohort study. J Endocr Soc. https://doi.org/10.1210/jendso/bvac045
-
Clayton RN, Jones PW, Reulen RC et al (2016) Mortality in patients with Cushing’s disease more than 10 years after remission: a multicentre, multinational, retrospective cohort study. Lancet Diabetes Endocrinol 4(7):569–576. https://doi.org/10.1016/S2213-8587(16)30005-5
-
Nieman LK, Biller BM, Findling JW et al (2008) The diagnosis of Cushing’s syndrome: an endocrine society clinical practice guideline. J ClinEndocrinolMetab 93(5):1526–1540. https://doi.org/10.1210/jc.2008-0125
-
Petersenn S, Newell-Price J, Findling JW, Gu F, Maldonado M, Sen K, Salgado LR, Colao A, Biller BM, Pasireotide B2305 Study Group (2014) High variability in baseline urinary free cortisol values in patients with Cushing’s disease. ClinEndocrinol 80(2):261–9. https://doi.org/10.1111/cen.12259
-
Sandouk Z, Johnston P, Bunch D, Wang S, Bena J, Hamrahian A, Kennedy L (2018) Variability of late-night salivary cortisol in cushing disease: a prospective study. J ClinEndocrinolMetab 103(3):983–990. https://doi.org/10.1210/jc.2017-02020
-
Barbot M, Trementino L, Zilio M et al (2016) Second-line tests in the differential diagnosis of ACTH-dependent Cushing’s syndrome. Pituitary 19(5):488–495. https://doi.org/10.1007/s11102-016-0729-y
-
Barbot M, Albiger N, Koutroumpi S et al (2013) Predicting late recurrence in surgically treated patients with Cushing’s disease. ClinEndocrinol (Oxf) 79(3):394–401. https://doi.org/10.1111/cen.12133
-
Reincke M, Albani A, Assie G et al (2021) Corticotrophtumor progression after bilateral adrenalectomy (Nelson’s syndrome): systematic review and expert consensus recommendations. Eur J Endocrinol 184(3):P1–P16. https://doi.org/10.1530/EJE-20-1088
-
Williams B, Mancia G, Spiering W, AgabitiRosei E, Azizi M, Burnier M, Clement DL, Coca A, de Simone G, Dominiczak A, Kahan T, Mahfoud F, Redon J, Ruilope L, Zanchetti A, Kerins M, Kjeldsen SE, Kreutz R, Laurent S, Lip GYH, McManus R, Narkiewicz K, Ruschitzka F, Schmieder RE, EvgenyShlyakhto CT, Aboyans V, Desormais L, ESC Scientific Document Group (2018) ESC/ESH Guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the european society of cardiology (ESC) and the european society of hypertension (ESH). Eur Heart J 39(33):3021–3104. https://doi.org/10.1093/eurheartj/ehy339
-
American Diabetes Association Professional Practice Committee (2022) Classification and diagnosis of diabetes: standards of medical care in diabetes-2022. Diabetes Care. 45(1):S17–S38. https://doi.org/10.2337/dc22-S002
-
Yumuk V, Tsigos C, Fried M, Schindler K, Busetto L, Micic D, Toplak H, Obesity Management Task Force of the European Association for the Study of Obesity (2015) European guidelines for obesity management in adults. Obes Facts 8(6):402–24. https://doi.org/10.1159/000442721
-
Expert Panel on Detection Evaluation and Treatment of High Blood Cholesterol in Adults (2001) Executive summary of the third report of the national cholesterol education program (NCEP) expert panel on detection evaluation and treatment of high blood cholesterol in adults (Adult Treatment Panel III). JAMA. 285(19):2486–2497. https://doi.org/10.1001/jama.285.19.2486
-
Mach F, Baigent C, Catapano AL et al (2019) ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 41(1):111–188. https://doi.org/10.1093/eurheartj/ehz455
-
Fleseriu M, Hashim IA, Karavitaki N, Melmed S, Murad MH, Salvatori R, Samuels MH (2016) Hormonal replacement in hypopituitarism in adults: an endocrine society clinical practice guideline. J ClinEndocrinolMetab 101(11):3888–3921. https://doi.org/10.1210/jc.2016-2118
-
Rubinstein G, Osswald A, Hoster E, Losa M, Elenkova A, Zacharieva S, Machado MC, Hanzu FA, Zopp S, Ritzel K, Riester A, Braun LT, Kreitschmann-Andermahr I, Storr HL, Bansal P, Barahona MJ, Cosaro E, Dogansen SC, Johnston PC, Santos de Oliveira R, Raftopoulos C, Scaroni C, Valassi E, van der Werff SJA, Schopohl J, Beuschlein F, Reincke M (2020) Time to diagnosis in cushing’s syndrome: a meta-analysis based on 5367 patients. J ClinEndocrinolMetab 105(3):12. https://doi.org/10.1210/clinem/dgz136
-
BreslowNE DNE (1987) Statistical methods in cancer research. Volume II–The design and analysis of cohort studies. OxfordUniversity Press, New York
-
Mondin A, Ceccato F, Voltan G et al (2023) Treatment complications and mortality of Cushing’s disease: report on data collected over a 20-year period at a referral centre. EJEA. https://doi.org/10.1530/endoabs.90.P416
-
Clayton RN, Raskauskiene D, Reulen RC, Jones PW (2011) Mortality and morbidity in Cushing’s disease over 50 years in Stoke-on-Trent, UK: audit and meta-analysis of literature. J ClinEndocrinolMetab 96(3):632–642. https://doi.org/10.1210/jc.2010-1942
-
Lambert JK, Goldberg L, Fayngold S, Kostadinov J, Post KD, Geer EB (2013) Predictors of mortality and long-term outcomes in treated Cushing’s disease: a study of 346 patients. J ClinEndocrinolMetab 98(3):1022–1030. https://doi.org/10.1210/jc.2012-2893
-
Sharma ST, Nieman LK, Feelders RA (2015) Comorbidities in Cushing’s disease. Pituitary 18(2):188–194. https://doi.org/10.1007/s11102-015-0645-6.PMID:25724314;PMCID:PMC4374115
-
Schernthaner-Reiter MH, Siess C, Gessl A et al (2019) Factors predicting long-term comorbidities in patients with Cushing’s syndrome in remission. Endocrine. 64(1):157–168. https://doi.org/10.1007/s12020-018-1819-6
-
Visseren FLJ, Mach F, Smulders YM et al (2022) 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice: developed by the task Force for cardiovascular disease prevention in clinical practice with representatives of the european society of cardiology and 12 medical societies with the special contribution of the european association of preventive cardiology (EAPC). Rev EspCardiol 75(5):429. https://doi.org/10.1016/j.rec.2022.04.003
-
Papakokkinou E, Olsson DS, Chantzichristos D, Dahlqvist P, Segerstedt E, Olsson T, Petersson M, Berinder K, Bensing S, Höybye C, Edén-Engström B, Burman P, Bonelli L, Follin C, Petranek D, Erfurth EM, Wahlberg J, Ekman B, Åkerman AK, Schwarcz E, Bryngelsson IL, Johannsson G, Ragnarsson O (2020) Excess morbidity persists in patients with cushing’s disease during long-term remission: a swedish nationwide study. J ClinEndocrinolMetab. 105(8):291. https://doi.org/10.1210/clinem/dgaa291
-
Barbot M, Daidone V, Zilio M, Albiger N, Mazzai L, Sartori MT, Frigo AC, Scanarini M, Denaro L, Boscaro M, Casonato S, Ceccato F, Scaroni C (2015) Perioperative thromboprophylaxis in Cushing’s disease: what we did and what weare doing? Pituitary 18(4):487–493. https://doi.org/10.1007/s11102-014-0600-y
-
Barbot M, Ceccato F, Scaroni C (2018) Diabetes mellitus secondary to cushing’s disease. Front Endocrinol 5(9):284. https://doi.org/10.3389/fendo.2018.00284
-
Lacroix A, Feelders RA, StratakisCA NLK (2015) Cushing’s syndrome. Lancet 386(9996):913–927. https://doi.org/10.1016/S0140-6736(14)61375-1
-
Perez-Vega C, Ramos-Fresnedo A, Tripathi S, Domingo RA, Ravindran K, Almeida JP, Peterson J, Trifiletti DM, Chaichana KL, Quinones-Hinojosa A, Samson SL (2022) Treatment of recurrent and persistent Cushing’s disease after first transsphenoidal surgery: lessons learned from an international meta-analysis. Pituitary 25(3):540–549. https://doi.org/10.1007/s11102-022-01215-1
-
Valassi E, Franz H, Brue T, Feelders RA, Netea-Maier R, Tsagarakis S, Webb SM, Yaneva M, Reincke M, Droste M, Komerdus I, Maiter D, Kastelan D, Chanson P, Pfeifer M, Strasburger CJ, Tóth M, Chabre O, Krsek M, Fajardo C, Bolanowski M, Santos A, Trainer PJ, Wass JAH, Tabarin A, ERCUSYN Study Group (2018) Preoperative medical treatment in Cushing’s syndrome: frequency of use and its impact on postoperative assessment: data from ERCUSYN. Eur J Endocrinol 178(4):399–409. https://doi.org/10.1530/EJE-17-0997
-
Barbot M, Albiger N, Ceccato F, Zilio M, Frigo AC, Denaro L, Mantero F, Scaroni C (2014) Combination therapy for Cushing’s disease: effectiveness of two schedules of treatment: should we start with cabergoline or ketoconazole? Pituitary 17(2):109–117. https://doi.org/10.1007/s11102-013-0475-3)
-
van der Pas R, de Bruin C, Pereira AM, Romijn JA, Netea-Maier RT, Hermus AR, Zelissen PM, de Jong FH, van der Lely AJ, de Herder WW, Webb SM, Lamberts SW, Hofland LJ, Feelders RA (2013) Cortisol diurnal rhythm and quality of life after successful medical treatment of Cushing’s disease. Pituitary 16(4):536–544. https://doi.org/10.1007/s11102-012-0452-2
-
Findling JW, Fleseriu M, Newell-Price J, Petersenn S, Pivonello R, Kandra A, Pedroncelli AM, Biller BM (2016) Late-night salivary cortisol may be valuable for assessing treatment response in patients with Cushing’s disease: 12-month. Phase III Pasireotide Study Endocrine 54(2):516–523. https://doi.org/10.1007/s12020-016-0978-6
-
Newell-Price J, Pivonello R, Tabarin A, Fleseriu M, Witek P, Gadelha MR et al (2020) Use of late-night salivary cortisol to monitor response to medical treatment in Cushing’s disease. Eur J Endocrinol 182(2):207–17
-
Bornstein SR, Allolio B, Arlt W et al (2016) Diagnosis and treatment of primary adrenal insufficiency: an endocrine society clinical practice guideline. J ClinEndocrinolMetab 101(2):364–389. https://doi.org/10.1210/jc.2015-1710
-
Hakami OA, Ahmed S, Karavitaki N (2021) Epidemiology and mortality of Cushing’s syndrome. Best Pract Res Clin Endocrinol Metab 35(1):101521. https://doi.org/10.1016/j.beem.2021.101521
-
Albiger NM, Occhi G, Sanguin F, Iacobone M, Casarrubea G, Ferasin S, Mantero F, Scaroni C (2011) Adrenal nodules in patients with Cushing’s disease: prevalence, clinical significance and follow-up. J Endocrinol Invest 34(8):e204–e209. https://doi.org/10.3275/7349
-
Di Dalmazi G, Timmers HJLM, Arnaldi G et al (2019) Somatic PRKACA mutations: association with transition from pituitary-dependent to adrenal-dependent cushing syndrome. J ClinEndocrinolMetab 104(11):5651–5657. https://doi.org/10.1210/jc.2018-02209
Funding
Open access funding provided by Università degli Studi di Padova within the CRUI-CARE Agreement. The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics declarations
Competing interests
Authors certify that they have no affiliations with or involvement in any organization or entity with any financial or non-financial interest in the subject matters discussed in this manuscript.
Ethical approval
The current study was designed in accordance with the principles of the Declaration of Helsinki and approved by the Ethical Committee of the province of Padova (protocol code 236n/AO/22, date of approval 29 April 2022).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
-
1
-
-


YOU’RE INVITED! A4M/Metabolic Medical Institute (MMI) Webinar on Updates on Treating Hypothyroidism
Dr. Theodore Friedman (The Wiz) will giving a webinar on Updates on Treating Hypothyroidismregister at https://us02web.zoom.us/Topics to be discussed include:
- New articles showing patients prefer desiccated thyroid
- Moving away from a TSH-centered approach
- New thyroid hormone preparations, including Adthyza
-
•Does biotin affect thyroid tests?
•Hypothyroidism diet? - What is the difference between desiccated thyroid and synthetic thyroid hormones?
- Is rT3 important?
Wednesday • August 2, 2023 • 4 PM PDTwebinar/register/WN_ kRyqZFlrSgKj54CuK7OQqQ
Slides will be available on the day of the talk here.
There will be plenty of time for questions using the chat button.
For more information, email us at mail@goodhormonehealth.com-
1
-

The most common procedure to remove pituitary tumors is transsphenoidal adenomectomy. It allows the removal of the tumor with minimal damage to the surrounding structures. The surgical team accesses the pituitary gland through the sphenoid sinus — a hollow space behind the nasal passages and below the pituitary gland.
If performed in specialized centers and by an experienced pituitary surgeon, this type of surgery is reported to result in an overall cure rate, or full remission, of Cushing’s disease for 80% to 90% of patients. A higher success rate is seen with smaller tumors.
However, reported remission rates vary considerably, mainly due to differences in the criteria used to define disease remission.
In some cases, a second transsphenoidal adenomectomy is required to fully remove tumor tissue; in others, the initial surgical procedure is paired with a second form of treatment, such as radiation therapy or certain medications.
Given the complexity of the procedure, the guidelines recommend patients undergo surgery in specialized Pituitary Tumor Centers of Excellence. Patients also are advised to have the surgery performed by an experienced pituitary neurosurgeon.
Follow-up for all patients should be conducted by a multidisciplinary team, including a pituitary endocrinologist.
Lifelong monitoring for disease recurrence is required.
-
1
-
-
Abstract
Purpose. Few related factors of low bone mass in Cushing’s disease (CD) have been identified so far, and relevant sufficient powered studies in CD patients are rare. On account of the scarcity of data, we performed a well-powered study to identify related factors associated with low bone mass in young CD patients.
Methods. This retrospective study included 153 CD patients (33 males and 120 females, under the age of 50 for men and premenopausal women). Bone mineral density (BMD) of the left hip and lumbar spine was measured by dual energy X-ray absorptiometry (DEXA). In this study, low bone mass was defined when the Z score was −2.0 or lower. Results. Among those CD patients, low bone mass occurred in 74 patients (48.37%). Compared to patients with normal BMD, those patients with low bone mass had a higher level of serum cortisol at midnight (22.31 (17.95-29.62) vs. 17.80 (13.75-22.77), ), testosterone in women (2.10 (1.33–2.89) vs. 1.54 (0.97–2.05), ), higher portion of male (32.43% vs. 11.54%, ) as well as hypertension (76.12% vs. 51.67%, ), and lower IGF-1 index (0.59 (0.43–0.76) vs. 0.79 (0.60–1.02), ). The Z score was positively associated with the IGF-1 index in both the lumbar spine (r = 0.35153, ) and the femoral neck (r = 0.24418, ). The Z score in the femoral neck was negatively associated with osteocalcin (r = −0.22744, ). Compared to the lowest tertile of the IGF-1 index (<0.5563), the patients with the highest tertile of the IGF-1 index (≥0.7993) had a lower prevalence of low bone mass (95% CI 0.02 (0.001–0.50), ), even after adjusting for confounders such as age, gender, duration, BMI, hypertension, serum cortisol at midnight, PTH, and osteocalcin.
Conclusions. The higher IGF-1 index was independently associated with lower prevalence of low bone mass in young CD patients, and IGF-1 might play an important role in the pathogenesis of CD-caused low bone mass.
1. Introduction
Cushing’s disease (CD), caused by an adrenocorticotropic hormone (ACTH)-secreting pituitary tumor, is a rare disease with approximately 1.2 to 2.4 new cases per million people each year [1].
Osteoporosis has been recognized as a serious consequence of endogenous hypercortisolism since the first description in 1932 [2]. The prevalence of osteoporosis is around 38–50%, and the rate of atraumatic compression fractures is 15.8% in CD patients [3]. After cortisol normalization and appropriate treatment, recovery of the bone impairment occurs slowly (6–9 years) and partially [4, 5]. Hypercortisolemia impairs bone quality through multiple mechanisms [6]. Growth hormone (GH) and insulin-like growth factor 1 (IGF-1) play a crucial role in bone growth and development [7]. IGF-1 is considered essential for the longitudinal growth of bone, skeletal maturity, and bone mass acquisition not only during growth but also in the maintenance of bone in adults [8]. Previous research studies revealed that low serum IGF-1 levels were associated with a 40% increased risk of fractures [9, 10], and serum IGF-1 levels could be clinically useful for evaluating the risk of spinal fractures [11]. In Marl Hotta’s research, extremely low or no response of plasma GH to recombinant human growth hormone (hGRH) injection was noted in CD patients. This result suggested that the diminished hGRH-induced GH secretion in patients with Cushing’s syndrome might be caused by the prolonged period of hypercortisolemia [12]. Other surveys indicated that glucocorticoids, suppressing GH–IGF-1 and the hypothalamic-pituitary-gonadal axes, lead to decreased number and dysfunction of osteoblast [13].
However, the exact mechanism is still unclear, and few risk factors for osteoporosis in CD have been identified so far. Until now, relevant and sufficiently powered studies in CD patients have been rare [14, 15]. Early recognition of the changes in bone mass in CD patients contributes to early diagnosis of bone mass loss and prompt treatment, which could help minimize the incidence of adverse events such as fractures.
On account of the scarcity of data and pressing open questions concerning risk evaluation and management of osteoporosis, we performed a well-powered study to identify the related factors associated with low bone mass in young CD patients at the time of diagnosis.
2. Materials and Methods
2.1. Subjects
This retrospective study enrolled 153 CD patients (33 males and 120 females) from the Department of Endocrinology and Metabolism of Huashan Hospital between January 2010 and February 2021. All subjects were evaluated by the same group of endocrinologists for detailed clinical evaluation. This study, which was in complete adherence to the Declaration of Helsinki, was approved by the Human Investigation Ethics Committee at Huashan Hospital, Fudan University (No. 2017M011). We collected data on demographic characteristics, laboratory tests, and bone mineral density.
Inclusion criteria included the following: (1) willingness to participate in the study; (2) premenopausal women ≥18 years old, men ≥18 years old but younger than 50 years old, and young women (<50 years old) with menstrual abnormalities who were associated with CD after excluding menstrual abnormalities caused by other causes; (3) diagnosis of CD according to the updated diagnostic criteria [16]; and (4) pathological confirmation after transsphenoidal surgery (positive immunochemistry staining with ACTH). Exclusion criteria included Cushing’s syndrome other than pituitary origin.
2.2. Clinical and Biochemical Methods
IGF-1 was measured using the Immulite 2000 enzyme-labeled chemiluminescent assay (Siemens Healthcare Diagnostic, Surrey, UK). Other endocrine hormones, including cortisol (F), 24-hour urinary free cortisol (24hUFC), adrenocorticotropic hormone (ACTH), prolactin (PRL), luteinizing hormone (LH), follicle stimulating hormone (FSH), estrogen (E2), progesterone (P), testosterone (T), thyroid stimulating hormone (TSH), and free thyroxine (FT4), were carried out by the chemiluminescence assay (Advia Centaur CP). Intra-assay and interassay coefficients of variation were less than 8 and 10%, respectively, for the estimation of all hormones.
Bone metabolism markers included osteocalcin (OC), type I procollagen amino-terminal peptide (P1NP), parathyroid hormone (PTH), and 25-hydroxyvitamin D (25(OH)VD), measured in a Roche Cobas e411 analyzer using immunometric assays (Roche Diagnostics, Indianapolis, IN, USA).
The IGF-1 index was defined as the ratio of the measured value to the respective upper limit of the reference range for age and sex. Body mass index (BMI) was calculated using the following formula: weight (kg)/height2 (m2). The bone mineral density (BMD) measuring instrument was Discovery type W dual energy X-ray absorptiometry from the American HOLOGIC company. Quality control tests were conducted every working day. Before examination, the date of birth, height, weight, and menopause date of the examiner were accurately recorded, and then BMD (g/cm2) of the left hip and lumbar spine were measured by DEXA. Z value was used for premenopausal women and men younger than 50 years old, and Z-value = (measured value − mean bone mineral density of peers)/standard deviation of BMD of peers [17, 18]. In this study, low bone mass was defined as a Z-value of −2.0 or lower.
2.3. Statistical Analysis
The baseline characteristics were compared between CD patients with and without low bone mass by using the Student’s t-test for continuous variables and the χ2 test for category variables. Bone turnover markers, alanine aminotransferase (ALT), triglyceride (TG), IGF-1 index, thyroid stimulating hormone (TSH), free triiodothyronine (FT3), free thyroxine (FT4), testosterone (T), 24 hours of urine cortisol (24 h UFC), and serum cortisol at 8 a.m. (F8 am) and at midnight (F24 pm) were not in normal distribution, so variables mentioned above were Log10-transformed, which could be used as continuous variables during statistical analysis. Participants were categorized into three groups according to tertiles of the IGF-1 index: <0.5986, 0.5986–0.8380, and >0.8380. The linear trend across IGF-1 index tertiles was tested using linear regression analysis for continuous variables and the Cochran–Armitage test for categorical variables. We used a multivariate logistic regression model to identify related factors that are independently associated with the risk of low bone mass. Variables included in the multivariate logistic regression model were selected based on the Spearman rank correlation analysis and established traditional low bone mass risk factors as priors. The results were presented as odds ratios (OR) and the corresponding 95% confidence intervals (CI). Significance tests were two-tailed, with value <0.05 considered statistically significant for all analyses. Statistical analysis was performed using SAS version 9.3 (SAS Institute Inc, Cary, NC, USA).
3. Results
3.1. The Prevalence of Low Bone Mass in Young Cushing’s Disease Patients
From the inpatient system of Huashan hospital, a total of 153 CD patients under the age of 50 for men and premenopausal women (some with menstrual abnormalities were associated with CD) were included, aged from 13 to 49 years, with an average age of 34.25 ± 8.39 years. There were 33 males (21.57%) and 120 females (78.43%). These CD patients included newly diagnosed CD, recurrences of CD, and CD without remission after treatment. There were no differences in the prevalence of different statuses of CD between the two groups (Table 1).
Among these CD patients, low bone mass occurred in 74 patients (48.37%), including 24 men and 50 women. The prevalence of low bone mass was 41.67% and 72.73% in female and male CD patients, respectively, and 42 (56.76%) patients suffered from low bone mass in the lumbar spine only, while 10 (13.51%) patients had low bone mass in the femoral neck only, and 22 (29.73%) patients had low bone mass in both parts.
In female patients with low bone mass, 27 (54%) had low bone mass in the lumbar region only, 9 (18%) in the femoral neck only, and 14 (28%) had low bone mass in both parts. For male patients with low bone mass, 16 (66.67%) patients had low bone mass only in the lumbar region, and the rest (8, 33.33%) had low bone mass in both parts.
Ten patients had a history of fragility fractures (6 ribs, 3 vertebrae, 1 femoral neck, and ribs), and all of them achieved low bone mass in BMD.
3.2. Baseline Characteristics of Cushing’s Disease Patients with and without Low Bone Mass
These CD patients were divided into two groups with and without low bone mass (Table 1). Compared to patients without low bone mass, those low bone mass patients had a higher level of diastolic blood pressure (DBP) (97.07 ± 13.69 vs. 89.76 ± 13.43, ), serum creatinine (66.15 ± 24.33 vs. 55.90 ± 13.35, ), uric acid (0.36 ± 0.10 vs. 0.32 ± 0.10, ), cholesterol (5.57 ± 1.30 vs. 5.06 ± 1.47, ), testosterone in women (2.10 (1.33–2.89) vs. 1.54 (0.97–2.05), ), F24 pm (22.31 (17.95–29.62) vs. 17.80 (13.75–22.77), ), and higher portion of male (32.43% vs. 11.54%, ), as well as hypertension (76.12% vs. 51.67%, ). The low bone mass group had a lower IGF-1 index (0.59 (0.43–0.76) vs. 0.79 (0.60–1.02), ) and FT3 level (3.54 (3.16–4.04) vs. 3.98 (3.47–4.45), ) than those without low bone mass. CD patients without low bone mass were more likely to have serum IGF-1 above the upper limit of the normal reference range (ULN) with age-adjusted (18, 26.87% vs. 3, 4.84%, ). No differences of bone turnover makers were found between the two groups.
3.3. Association between Baseline Characteristics and BMD
Spearman’s rank correlation analysis was used to explore the related factors of low bone mass in young CD patients (Table 2). The results indicated that the Z score in the lumbar spine was positively associated with age at diagnosis (r = 0.18801, ), IGF-1 index (r = 0.35153, ), FT3 level (r = 0.24117, ), estradiol in women (r = 0.2361, ), and occurrence of normal menstruation in females (r = 0.2267, ). Meanwhile, SBP (r = −0.21575, ), DBP (r = −0.32538, ), ALT (r = −0.17477, ), serum creatinine (r = −0.36072, ), cholesterol (r = −0.20205, ), testosterone in women (r = −0.2700, ), F8 am (r = −0.18998, ), and serum cortisol at midnight (r = −0.27273, ) were negatively associated with the Z-score in the lumbar spine. The results also illustrated that the Z-score in the femoral neck was positively associated with BMI (r = 0.33926, ), IGF-1 index (r = 0.24418, ), FT3 level (r = 0.20487, ), and occurrence of normal menstruation in females (r = 0.2393, ). Serum creatinine (r = −0.1932, ), osteocalcin (r = −0.22744, ), and testosterone in women (r = −0.2363, ) were negatively associated with the Z-score in the femoral neck.
3.4. IGF-1 Index and Low Bone Mass
Participants were categorized into the following three groups according to tertiles of the preoperative IGF-1 index: <0.5986 (tertiles 1), 0.5986–0.8380 (tertiles 2), and >0.8380 (tertiles 3). With the IGF-1 index increasing, the level of PTH decreased (54.85 (38.35–66.2), 38.9 (26.6–66.9), 36 (25.5–47.05), and ), while other bone metabolism makers, including PINP, osteocalcin, and 25 (OH) VD, showed no differences among the three groups (Figures 1(a)–1(d)). With the increase in the IGF-1 index level, the Z-score of both vertebra lumbalis (tertiles 1: −2.4 (−3.3∼−1.5); tertiles 2: −1.9 (−2.3∼−1.0); tertiles 3: −1.15 (−1.9∼−0.4), ) and the neck of femur (tertiles 1: −1.7 (−2.3∼−0.95); tertiles 2: −1.2 (−1.9∼−0.5); tertiles 3: −1.0 (−1.5∼−0.5), ) increased gradually (Figures 2(a) and 2(b)). Meanwhile, prevalence of low bone mass decreased (68.29%, 53.33%, 23.81%, ) (Figure 3(a)) both in the vertebra lumbalis (63.41%, 48.89%, 16.67%, ) and the neck of femur (32.5%, 11.11%, 11.19%, ), with the increasing of the IGF-1 index level (Figures 3(b) and 3(c)).
Bone turnover makers in three groups according to tertiles of the preoperative IGF-1 index. Tertiles 1: <0.5986, tertiles 2: 0.5986–0.8380, and tertiles 3 >0.8380. a for PINP; b for osteocalcin; c for PTH; d for VD-OH25. (a) p for trend = 0.2601. (b) p for trend = 0.1310. (c) p for trend = 0.008. (d) p for trend = 0.7956.Prevalence of low bone mass according to tertiles of the preoperative IGF-1 index. With increment of the IGF-1 index level, prevalence of low bone mass decreased, both in the vertebra lumbalis and neck of femur. Tertiles 1: <0.5986, tertiles 2: 0.5986–0.8380, and tertiles 3 >0.8380. (a) p for trend = 0.0002. (b) p for trend = 0.0169. (c) p for trend < 0.0001.In the logistic regression analysis of the related factors of low bone mass, most of the potentially relevant factors were put into this model; only the IGF-1 index was still significantly negatively associated with the prevalence of low bone mass after adjusting for covariables. The results indicated that compared to the patients in the lowest tertile of the IGF-1 index (<0.5563), those with the highest tertile of the IGF-1 index (≥0.7993) had a lower prevalence of low bone mass (95% CI 0.16 (0.06–0.41), ). After adjusting for age, gender, and BMI, the patients in the highest tertile of the IGF-1 index still conferred a lower prevalence of low bone mass (95% CI 0.15 (0.06–0.42), ). The association between the IGF-1 index and low bone mass still existed (95% CI 0.02 (0.001–0.5), ) even after adjusting for age, gender, CD duration, BMI, hypertension, dyslipidemia, diabetes, ALT, Scr, FT3, F24 pm, PTH, and osteocalcin (Table 3). In comparison to the reference population, the participants in the middle tertile of the IGF-1 index (0.5563–0.7993) had no different risk of low bone mass.
4. Discussion
Our results revealed that low bone mass occurred in around half of young CD patients, affecting more males than females, and mostly in the lumbar spine. The CD patients in our study had a high prevalence (48.37%) of low bone mass at the baseline. This was in accordance with the findings of previous research, and the reported prevalence of osteoporosis due to excess endogenous cortisol ranges from 22% to 59% [19–25]. In this study, CD patients’ lumbar vertebrae were more severely affected than the neck of the femur. It is reported that lumbar vertebrae, containing more trabecular bone than femur neck, were more vulnerable to endogenous cortisol [26].
Our results also indicated that men were more prone to low bone mass than women in CD, which was in accordance with several other studies [23, 27, 28]; possibly, the deleterious effect of cortisol excess on BMD might overrule the protective effects of sex hormones, and men were more often hypogonadal compared with women in CD patients. In our study, patients with low bone mass had a significantly higher level of F24 pm. Both cortisol levels in the morning and at midnight, were negatively associated with the Z-score of BMD in the lumbar spine at diagnosis. But these results were not seen in the femoral neck at diagnosis. This further indicated that lumbar vertebrae were more vulnerable to endogenous cortisol. BMI was considered to be associated with bone mass [29]. In our study, higher BMI was associated with higher BMD at diagnosis in the femur neck but not in the lumbar vertebrae, consistent with other studies [30].
Interestingly, besides the above known related factors, we also found that a higher level of the IGF-1 index was strongly associated with a lower prevalence of low bone mass, both in the vertebra lumbalis and the neck of the femur, independently of age, gender, duration, BMI, hypertension, dyslipidemia, diabetes, level of ALT, creatinine, FT3, and F24 pm. The IGF-1 index was also positively associated with the BMD Z-score, both in the lumbar spine and the femoral neck. So far, there have been few studies concerning the association between IGF-1 and low bone mass in Cushing’s disease patients. As we know, GH [31, 32] and IGF-1 [33] have been demonstrated to increase both bone formation (e.g., collagen synthesis) and bone resorption. However, in CD patients, glucocorticoids resulted in decreased number and dysfunction of osteoblasts by inhibiting GH-IGF-1 axes [34, 35]. In vitro studies suggested that at high concentrations of glucocorticoids, a decreased release of GHRH had been reported [36–38]; therefore, GH-IGF-1 axes were inhibited. IGF-1 possessed anabolic mitogenic actions in osteoblasts while reducing the anabolic actions of TGF-β [39]. The decrease in IGF-1 might be a risk factor for low bone mass in CD patients. In vitro studies had also indicated that the suppressive effects of glucocorticoids on osteoblast function can be partially reversed by GH or IGF treatment [8]. In recent years, some studies have also shown that patients with untreated Cushing’s disease may have elevated IGF-1, and mildly elevated IGF-1 in Cushing’s disease does not imply pathological growth hormone excess. Higher IGF-1 levels could predict better outcomes in CD [40, 41]. Possible mechanisms were not clear, which might involve changes in IGF binding proteins (IGFBPs), interference in IGFBP fragments, IGF-1 synthesis or clearance, and/or the effects of hyperinsulinism induced by excess glucocorticoids. In our study, the results also showed that IGF-1 was an independent protective factor for low bone mass in CD patients.
Our study was one of the few well-powered research studies on the association of IGF-1 levels with low bone mass in young CD patients. These represented important strengths of our study, especially given the rarity of CD. The main limitation of this study was its retrospective nature. This could not prove causality. A prospective study should be conducted to explore the causality between IGF-1 and osteoporosis in CD patients. In addition, this study lacked morphometric data for spinal fractures in all patients, which may underestimate the incidence of fractures and osteoporosis. However, our study indicated that a lower IGF-1 index level was significantly associated with low bone mass in young CD patients, which might provide a new aspect to understand the possible risk factors and mechanism of osteoporosis in CD patients.
In conclusion, our study found that a higher IGF-1 index was independently and significantly associated with decreased prevalence of low bone mass in young CD patients, drawing attention to the role of IGF-1 in the pathogenesis of CD-caused low bone mass and may support the exploration of this pathway in therapeutic agent development in antiosteoporosis in CD.
Data Availability
The data used to support the findings of the study are available on request from the authors.
Additional Points
Through a retrospective study of a large sample of Cushing’s disease (CD) patients from a single center, we found that a higher IGF-1 index was independently associated with a lower prevalence of low bone mass in young CD patients and IGF-1 might play an important role in the pathogenesis of CD-caused low bone mass.
Disclosure
Wanwan Sun and Quanya Sun were the co-first authors.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Authors’ Contributions
Wanwan Sun analyzed the data and wrote the manuscript. Quanya Sun collected the data. Hongying Ye and Shuo Zhang conducted the study design and quality control. All authors read and approved the final manuscript. Wanwan Sun and Quanya Sun contributed equally to this work.
Acknowledgments
The present study was supported by grants from the initial funding of the Huashan Hospital (2021QD023). The study was also supported by grants from Multidisciplinary Diagnosis and Treatment (MDT) demonstration project in research hospitals (Shanghai Medical College, Fudan University, no: DGF501053-2/014).
References
-
H. Nishioka and S. Yamada, “Cushing's disease,” Journal of Clinical Medicine, vol. 8, no. 11, p. 1951, 2019.
View at: Publisher Site | Google Scholar -
H. Cushing, “The basophil adenomas of the pituitary body and their clinical manifestations (pituitary basophilism),” Obesity Research, vol. 2, no. 5, pp. 486–508, 1994.
View at: Publisher Site | Google Scholar -
R. A. Feelders, S. J. Pulgar, A. Kempel, and A. M. Pereira, “Management of endocrine disease: the burden of Cushing's disease: clinical and health-related quality of life aspects,” European Journal of Endocrinology, vol. 167, no. 3, pp. 311–326, 2012.
View at: Publisher Site | Google Scholar -
R. Pivonello, M. De Leo, A. Cozzolino, and A. Colao, “The treatment of Cushing's disease,” Endocrine Reviews, vol. 36, no. 4, pp. 385–486, 2015.
View at: Publisher Site | Google Scholar -
R. Pivonello, M. C. De Martino, M. De Leo, C. Simeoli, and A. Colao, “Cushing's disease: the burden of illness,” Endocrine, vol. 56, no. 1, pp. 10–18, 2017.
View at: Publisher Site | Google Scholar -
R. S. Hardy, H. Zhou, M. J. Seibel, and M. S. Cooper, “Glucocorticoids and bone: consequences of endogenous and exogenous excess and replacement therapy,” Endocrine Reviews, vol. 39, no. 5, pp. 519–548, 2018.
View at: Publisher Site | Google Scholar -
R. Bouillon and A. Prodonova, “Growth hormone deficiency and peak bone mass: laboratory for experimental medicine and Endocrinology, catholic university of leuven, gasthuisberg, leuven, Belgium,” Journal of Pediatric Endocrinology and Metabolism, vol. 13, no. s2, pp. 1327–1342, 2000.
View at: Publisher Site | Google Scholar -
A. Giustina, G. Mazziotti, and E. Canalis, “Growth hormone, insulin-like growth factors, and the skeleton,” Endocrine Reviews, vol. 29, no. 5, pp. 535–559, 2008.
View at: Publisher Site | Google Scholar -
T. Sugimoto, K. Nishiyama, F. Kuribayashi, and K. Chihara, “Serum levels of insulin-like growth factor (IGF) I, IGF-binding protein (IGFBP)-2, and IGFBP-3 in osteoporotic patients with and without spinal fractures,” Journal of Bone and Mineral Research, vol. 12, no. 8, pp. 1272–1279, 1997.
View at: Publisher Site | Google Scholar -
P. Garnero, E. Sornay-Rendu, and P. D. Delmas, “Low serum IGF-1 and occurrence of osteoporotic fractures in postmenopausal women,” The Lancet, vol. 355, no. 9207, pp. 898-899, 2000.
View at: Publisher Site | Google Scholar -
C. Ohlsson, D. Mellström, D. Carlzon et al., “Older men with low serum IGF-1 have an increased risk of incident fractures: the MrOS Sweden study,” Journal of Bone and Mineral Research, vol. 26, no. 4, pp. 865–872, 2011.
View at: Publisher Site | Google Scholar -
M. Hotta, T. Shibasaki, A. Masuda et al., “Effect of human growth hormone-releasing hormone on GH secretion in Cushing's syndrome and non-endocrine disease patients treated with glucocorticoids,” Life Sciences, vol. 42, no. 9, pp. 979–984, 1988.
View at: Publisher Site | Google Scholar -
L. T. Braun and M. Reincke, “The effect of biochemical remission on bone metabolism in Cushing's syndrome: a 2-year follow-up study,” Journal of Bone and Mineral Research, vol. 36, no. 11, pp. 2281-2282, 2021.
View at: Publisher Site | Google Scholar -
I. Kanazawa, T. Yamaguchi, M. Yamamoto, M. Yamauchi, S. Yano, and T. Sugimoto, “Serum insulin-like growth factor-I level is associated with the presence of vertebral fractures in postmenopausal women with type 2 diabetes mellitus,” Osteoporosis International, vol. 18, no. 12, pp. 1675–1681, 2007.
View at: Publisher Site | Google Scholar -
A. Scillitani, G. Mazziotti, C. Di Somma et al., “Treatment of skeletal impairment in patients with endogenous hypercortisolism: when and how?” Osteoporosis International, vol. 25, no. 2, pp. 441–446, 2014.
View at: Publisher Site | Google Scholar -
M. Fleseriu, R. Auchus, I. Bancos et al., “Consensus on diagnosis and management of Cushing's disease: a guideline update,” Lancet Diabetes and Endocrinology, vol. 9, no. 12, pp. 847–875, 2021.
View at: Publisher Site | Google Scholar -
J. M. Liu, D. L. Zhu, Y. M. Mu, and W. B. Xia, “Chinese Society of Osteoporosis and Bone Mineral Research, the Chinese Society of Endocrinology, Chinese Diabetes Society, Chinese Medical Association; Chinese Endocrinologist Association, Chinese Medical Doctor Association,” Management of fracture risk in patients with diabetes-Chinese Expert Consensus Journal of Diabetes, vol. 11, pp. 906–919, 2019.
View at: Google Scholar -
P. M. Camacho, S. M. Petak, N. Binkley et al., “American association of clinical endocrinologists and American college of endocrinology clinical practice guidelines for the diagnosis and treatment of postmenopausal OSTEOPOROSIS-2020 update,” Endocrine Practice, vol. 26, no. 1, pp. 1–46, 2020.
View at: Publisher Site | Google Scholar -
C. V. dos Santos, L. Vieira Neto, M. Madeira et al., “Bone density and microarchitecture in endogenous hypercortisolism,” Clinical Endocrinology, vol. 83, no. 4, pp. 468–474, 2015.
View at: Publisher Site | Google Scholar -
N. Ohmori, K. Nomura, K. Ohmori, Y. Kato, T. Itoh, and K. Takano, “Osteoporosis is more prevalent in adrenal than in pituitary Cushing's syndrome,” Endocrine Journal, vol. 50, no. 1, pp. 1–7, 2003.
View at: Publisher Site | Google Scholar -
M. E. Randazzo, E. Grossrubatscher, P. Dalino Ciaramella, A. Vanzulli, and P. Loli, “Spontaneous recovery of bone mass after cure of endogenous hypercortisolism,” Pituitary, vol. 15, no. 2, pp. 193–201, 2012.
View at: Publisher Site | Google Scholar -
L. Tauchmanovà, R. Pivonello, C. Di Somma et al., “Bone demineralization and vertebral fractures in endogenous cortisol excess: role of disease etiology and gonadal status,” Journal of Clinical Endocrinology and Metabolism, vol. 91, no. 5, pp. 1779–1784, 2006.
View at: Publisher Site | Google Scholar -
E. Valassi, A. Santos, M. Yaneva et al., “The European Registry on Cushing's syndrome: 2-year experience. Baseline demographic and clinical characteristics,” European Journal of Endocrinology, vol. 165, no. 3, pp. 383–392, 2011.
View at: Publisher Site | Google Scholar -
L. Trementino, G. Appolloni, L. Ceccoli et al., “Bone complications in patients with Cushing's syndrome: looking for clinical, biochemical, and genetic determinants,” Osteoporosis International, vol. 25, no. 3, pp. 913–921, 2014.
View at: Publisher Site | Google Scholar -
A. W. van der Eerden, M. den Heijer, W. J. Oyen, and A. R. Hermus, “Cushing's syndrome and bone mineral density: lowest Z scores in young patients,” The Netherlands Journal of Medicine, vol. 65, no. 4, pp. 137–141, 2007.
View at: Google Scholar -
P. G. Lacativa and M. L. F. Farias, “Office practice of osteoporosis evaluation,” Arquivos Brasileiros de Endocrinologia and Metabologia, vol. 50, no. 4, pp. 674–684, 2006.
View at: Publisher Site | Google Scholar -
L. H. A. Broersen, F. M. van Haalen, T. Kienitz et al., “Sex differences in presentation but not in outcome for ACTH-dependent Cushing's syndrome,” Frontiers in Endocrinology, vol. 10, p. 580, 2019.
View at: Publisher Site | Google Scholar -
F. P. Giraldi, M. Moro, and F. Cavagnini, “Gender-related differences in the presentation and course of Cushing's disease,” Journal of Clinical Endocrinology and Metabolism, vol. 88, no. 4, pp. 1554–1558, 2003.
View at: Publisher Site | Google Scholar -
S. Morin, J. F. Tsang, and W. D. Leslie, “Weight and body mass index predict bone mineral density and fractures in women aged 40 to 59 years,” Osteoporosis International, vol. 20, no. 3, pp. 363–370, 2009.
View at: Publisher Site | Google Scholar -
M. Zilio, M. Barbot, F. Ceccato et al., “Diagnosis and complications of Cushing's disease: gender-related differences,” Clinical Endocrinology, vol. 80, no. 3, pp. 403–410, 2014.
View at: Publisher Site | Google Scholar -
G. Amato, C. Carella, S. Fazio, G. La Montagna, A. Cittadini, and D. Sabatini, “Body composition, bone metabolism, and heart structure and function in growth hormone (GH)-deficient adults before and after GH replacement therapy at low doses,” Journal of Clinical Endocrinology and Metabolism, vol. 77, no. 6, pp. 1671–1676, 1993.
View at: Publisher Site | Google Scholar -
S. A. Beshyah, E. Thomas, P. Kyd, P. Sharp, A. Fairney, and D. G. Johnston, “The effect of growth hormone replacement therapy in hypopituitary adults on calcium and bone metabolism,” Clinical Endocrinology, vol. 40, no. 3, pp. 383–391, 2010.
View at: Publisher Site | Google Scholar -
P. R. Ebeling, J. D. Jones, W. M. O'Fallon, C. H. Janes, and B. L. Riggs, “Short-term effects of recombinant human insulin-like growth factor I on bone turnover in normal women,” Journal of Clinical Endocrinology and Metabolism, vol. 77, no. 5, pp. 1384–1387, 1993.
View at: Publisher Site | Google Scholar -
G. Mazziotti and A. Giustina, “Glucocorticoids and the regulation of growth hormone secretion,” Nature Reviews Endocrinology, vol. 9, no. 5, pp. 265–276, 2013.
View at: Publisher Site | Google Scholar -
N. A. Tritos, “Growth hormone deficiency in adults with Cushing's disease,” Best Practice and Research Clinical Endocrinology and Metabolism, vol. 35, no. 2, Article ID 101474, 2021.
View at: Publisher Site | Google Scholar -
K. Nakagawa, T. Ishizuka, T. Obara, M. Matsubara, and K. Akikawa, “Dichotomic action of glucocorticoids on growth hormone secretion,” Acta Endocrinologica, vol. 116, no. 2, pp. 165–171, 1987.
View at: Publisher Site | Google Scholar -
G. Fernández-Vázquez, L. Cacicedo, M. J. Lorenzo, R. Tolón, J. López, and F. Sánchez-Franco, “Corticosterone modulates growth hormone-releasing factor and somatostatin in fetal rat hypothalamic cultures,” Neuroendocrinology, vol. 61, no. 1, pp. 31–35, 1995.
View at: Publisher Site | Google Scholar -
S. K. Fife, R. S. Brogan, A. Giustina, and W. B. Wehrenberg, “Immunocytochemical and molecular analysis of the effects of glucocorticoid treatment on the hypothalamic-somatotropic axis in the rat,” Neuroendocrinology, vol. 64, no. 2, pp. 131–138, 1996.
View at: Publisher Site | Google Scholar -
T. L. McCarthy, M. Centrella, and E. Canalis, “Cortisol inhibits the synthesis of insulin-like growth factor-I in skeletal cells,” Endocrinology, vol. 126, no. 3, pp. 1569–1575, 1990.
View at: Publisher Site | Google Scholar -
K. English, V. Chikani, G. Dimeski, and W. J. Inder, “Elevated insulin‐like growth factor‐1 in Cushing’s disease,” Clinical Endocrinology, vol. 91, no. 1, pp. 141–147, 2019.
View at: Publisher Site | Google Scholar -
E. Gezer, B. Çetinarslan, A. Selek et al., “The association between insulin-like growth factor 1 levels within reference range and early postoperative remission rate in patients with Cushing’s disease,” Endocrine Research, vol. 46, no. 3, pp. 92–98, 2021.
View at: Publisher Site | Google Scholar
Copyright
Copyright © 2023 Wanwan Sun et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.-
1
-
-
Abstract
Objectives
To assess the diagnostic performance of high-resolution contrast-enhanced MRI (hrMRI) with three-dimensional (3D) fast spin echo (FSE) sequence by comparison with conventional contrast-enhanced MRI (cMRI) and dynamic contrast-enhanced MRI (dMRI) with 2D FSE sequence for identifying pituitary microadenomas.
Methods
This single-institutional retrospective study included 69 consecutive patients with Cushing’s syndrome who underwent preoperative pituitary MRI, including cMRI, dMRI, and hrMRI, between January 2016 to December 2020. Reference standards were established by using all available imaging, clinical, surgical, and pathological resources. The diagnostic performance of cMRI, dMRI, and hrMRI for identifying pituitary microadenomas was independently evaluated by two experienced neuroradiologists. The area under the receiver operating characteristics curves (AUCs) were compared between protocols for each reader by using the DeLong test to assess the diagnostic performance for identifying pituitary microadenomas. The inter-observer agreement was assessed by using the κ analysis.
Results
The diagnostic performance of hrMRI (AUC, 0.95–0.97) was higher than cMRI (AUC, 0.74–0.75; p ≤ .002) and dMRI (AUC, 0.59–0.68; p ≤ .001) for identifying pituitary microadenomas. The sensitivity and specificity of hrMRI were 90–93% and 100%, respectively. There were 78% (18/23) to 82% (14/17) of the patients, who were misdiagnosed on cMRI and dMRI and correctly diagnosed on hrMRI. The inter-observer agreement for identifying pituitary microadenomas was moderate on cMRI (κ = 0.50), moderate on dMRI (κ = 0.57), and almost perfect on hrMRI (κ = 0.91), respectively.
Conclusions
The hrMRI showed higher diagnostic performance than cMRI and dMRI for identifying pituitary microadenomas in patients with Cushing’s syndrome.
Key Points
• The diagnostic performance of hrMRI was higher than cMRI and dMRI for identifying pituitary microadenomas in Cushing’s syndrome.
• About 80% of patients, who were misdiagnosed on cMRI and dMRI, were correctly diagnosed on hrMRI.
• The inter-observer agreement for identifying pituitary microadenomas was almost perfect on hrMRI.
Introduction
Cushing’s syndrome, caused by excessive exposure to glucocorticoids, is associated with considerable morbidity and increased mortality [1]. Cushing’s syndrome has diverse manifestations, including central obesity, moon facies, purple striae, and hypertension [2]. Cushing’s disease, due to adrenocorticotropic hormone (ACTH) hypersecretion from pituitary adenomas, is the most common etiology of ACTH-dependent Cushing’s syndrome [1, 2]. According to the Endocrine Society Clinical Practice Guideline, transsphenoidal surgery is the first-line treatment for Cushing’s disease [3]. The identification of pituitary adenomas on preoperative MRI can significantly increase the postoperative remission rate from 50 to 98% [4]. Therefore, it is critical to identify pituitary adenomas on MRI before surgery.
However, there are considerable challenges in identifying ACTH-secreting pituitary adenomas. This is because about 90% of the tumors are microadenomas (less than 10 mm in size) and the median diameter at surgery is about 5 mm [5, 6]. Conventional contrast-enhanced MRI (cMRI) using a two-dimensional (2D) fast spin echo (FSE) sequence has been routinely used to acquire images with 2- to 3-mm slice thickness, but some microadenomas are difficult to be identified on cMRI, resulting in false negatives reported in up to 50% of patients with Cushing’s disease [7]. Dynamic contrast-enhanced MRI (dMRI) increases the sensitivity of identifying pituitary adenomas to 66% [8], but it also increases false positives at the same time [9, 10]. The 3D spoiled gradient recalled (SPGR) sequence has been introduced in high-resolution contrast-enhanced MRI (hrMRI) to acquire images with 1- to 1.2-mm slice thickness. It is reported that the 3D SPGR sequence is superior to the 2D FSE sequence in the identification of pituitary adenomas with a sensitivity of up to 80% [11,12,13], but it cannot satisfy the clinical needs that about 20% of the lesions are still missed. Therefore, techniques are needed that can help better identify pituitary adenomas, particularly microadenomas. Previously, the 3D FSE sequence was recommended in patients with hyperprolactinemia [14]. Recently, the 3D FSE sequence has developed rapidly and can provide superior image quality with diminished artifacts [15]. Sartoretti et al demonstrated in a very effective fashion that the 3D FSE sequence is a reliable alternative for pituitary imaging in terms of image quality [16]. However, to our knowledge, few studies have investigated the diagnostic performance of 3D FSE sequences for identifying ACTH-secreting pituitary adenomas, particularly microadenomas.
The aim of our study was to assess the diagnostic performance of hrMRI with 3D FSE sequence by comparison with cMRI and dMRI with 2D FSE sequence for identifying ACTH-secreting pituitary microadenomas in patients with Cushing’s syndrome.
Materials and methods
This single-institutional retrospective study was approved by the Institutional Review Board of our hospital. The study was conducted in accordance with the Helsinki Declaration. The informed consent was waived due to the retrospective nature of the study.
Study participants
We retrospectively reviewed the medical records and imaging studies of 186 consecutive patients with ACTH-dependent Cushing’s syndrome, who underwent a combined protocol of cMRI, dMRI, and hrMRI from January 2016 to December 2020. Postoperative patients with Cushing’s disease (n = 97), patients with ectopic ACTH syndrome who underwent pituitary exploration (n = 2), and patients with macroadenomas (n = 5) or lack of pathology (n = 13) were excluded from the study. Finally, 69 patients with ACTH-dependent Cushing’s syndrome were included in the current study (Fig. 1) and the patients included were all surgically confirmed.
Fig. 1 
Flowchart of patient inclusion/exclusion process and image analysis. ACTH adrenocorticotropic hormone, CD Cushing’s disease, EAS ectopic ACTH syndrome, T1WI T1-weighted imaging, T2WI T2-weighted imaging
MRI protocol
All the patients were imaged on a 3.0 Tesla MR scanner (Discovery MR750w, GE Healthcare) using an 8-channel head coil. The MRI protocol included coronal T2-weighted imaging, coronal T1-weighted imaging, and sagittal T1-weighted imaging before contrast injection. After contrast injection of gadopentetate dimeglumine (Gd-DTPA) at 0.05 mmol/kg (0.1 mL/kg) with a flow rate of 2 mL/s followed by a 10-mL saline solution flush, dMRI and cMRI with 2D FSE sequence were obtained first, and hrMRI with 3D FSE sequence using variable flip angle technique was performed immediately afterward. Detailed acquisition parameters are presented in Table S1.
Image analysis: diagnostic performance
Image interpretation was independently conducted by two experienced neuroradiologists (F.F. and H.Y. with 25 and 16 years of experience in neuroradiology, respectively), who were blinded to patient information. The evaluation order of cMRI, dMRI, and hrMRI sequences was randomized. The identification of pituitary microadenomas on images was scored based on a three-point scale (0 = poor; 1 = fair; 2 = excellent). Scores of 1 or 2 represented the identification of the lesion. Reference standards were established by using all available imaging, clinical, surgical, and pathological resources, with a multidisciplinary team approach.
Image analysis: image quality
Two readers (Z.L. and B.H. with 4 years of experience in radiology, respectively) were asked to assess the image quality of cMRI, dMRI, and hrMRI. Before exposure to images used in the current study, these readers underwent a training session to make sure that they were comparable to the experienced neuroradiologists in terms of image quality assessment. Images were presented in a random order. Image quality was assessed by using a 5-point Likert scale [17], including overall image quality (1 = non-diagnostic; 2 = poor; 3 = fair; 4 = good; 5 = excellent), sharpness (1 = non-diagnostic; 2 = not sharp; 3 = a little sharp; 4 = moderately sharp; 5 = satisfyingly sharp), and structural conspicuity (1 = non-diagnostic; 2 = poor; 3 = fair; 4 = good; 5 = excellent). An example of image quality assessment is shown in Table S2. Final decision was made through a consensus agreement.
The mean signal intensity of pituitary microadenomas, pituitary gland, and noise on cMRI, dMRI, and hrMRI was measured using an operator-defined region of interest. For noise, a 10-mm2 region of interest was placed in the background, and noise was defined as the standard deviation of the signal intensity of the background [17]. For pituitary microadenomas and pituitary gland, the region of interest should include a representative portion of the structure. The mean signal intensity of the pituitary microadenoma was replaced with that of the pituitary gland when no microadenoma was identified. A signal-to-noise ratio (SNR) was defined as the mean signal intensity of the pituitary microadenoma divided by noise. A contrast-to-noise ratio (CNR) was defined as the absolute difference of the mean signal intensity between the normal pituitary gland and pituitary microadenomas divided by noise [17]. Supplementary Fig. 1 shows how to measure the SNR and CNR with the region of interest in a contrast-enhanced pituitary MRI. Supplementary Fig. 2 shows the selection of images for the SNR and CNR calculation.
Statistical analysis
The κ analysis was conducted to assess the inter-observer agreement for identifying pituitary microadenomas. The κ value was interpreted as follows: below 0.20, slight agreement; 0.21–0.40, fair agreement; 0.41–0.60, moderate agreement; 0.61–0.80, substantial agreement; greater than 0.80, almost perfect agreement.
To assess the diagnostic performance of cMRI, dMRI, and hrMRI for identifying pituitary microadenomas, the receiver operating characteristic curves were plotted and the area under curves (AUCs) were compared between MR protocols for each reader by using the DeLong test. Sensitivity, specificity, positive predictive value, and negative predictive value were calculated. The Mann–Whitney U test was used to evaluate the difference in image quality scores and the Wilcoxon signed-rank test was used to evaluate SNR and CNR measurements between MR protocols. A p value of less than 0.05 was considered statistically significant. Statistical analysis was performed using MedCalc Statistical Software (version 20.0.15; MedCalc Software) and SPSS Statistics (version 22.0; IBM).
Results
Clinical characteristics
A total of 69 patients (median age, 39 years; interquartile range [IQR], 29–54 years; 38 women [55%]) with ACTH-dependent Cushing’s syndrome were included in the study and their clinical characteristics are shown in Table 1. Among the 69 patients, 60 (87%) patients were diagnosed with Cushing’s disease and 9 (13%) were ectopic ACTH syndrome. The median disease course was 36 months (IQR, 12–78 months). The median serum cortisol, ACTH, and 24-h urine free cortisol level before surgery were 33.0 μg/dL (IQR, 25.1–40.1 μg/dL; normal range 4.0–22.3 μg/dL), 77.2 ng/L (IQR, 55.0–124.0 ng/L; normal range 0–46 ng/L), and 422.0 μg (IQR, 325.8–984.6 μg; normal range 12.3–103.5 μg), respectively. The median serum cortisol and 24-h urine free cortisol level after surgery were 3.0 μg/dL (IQR, 1.8–18.4 μg/dL) and 195.6 μg (IQR, 63.5–1240.3 μg), respectively. The median diameter of pituitary microadenomas was 5 mm (IQR, 4–5 mm), ranging from 3 to 9 mm.
Table 1 Clinical characteristics of the patients Diagnostic performance of cMRI, dMRI, and hrMRI for identifying pituitary microadenomas
The inter-observer agreement for identifying pituitary microadenomas by κ statistic between two readers was moderate on cMRI (κ = 0.50), moderate on dMRI (κ = 0.57), and almost perfect on hrMRI (κ = 0.91), respectively.
The diagnostic performance for identifying pituitary microadenomas on cMRI, dMRI, hrMRI, and combined cMRI and dMRI is summarized in Table 2. For reader 1, the diagnostic performance of hrMRI (AUC, 0.95; 95%CI: 0.87, 0.99) was higher than that of cMRI (AUC, 0.75; 95%CI: 0.63, 0.85; p = 0.002), dMRI (AUC, 0.59; 95%CI: 0.47, 0.71; p < 0.001), and combined cMRI and dMRI (AUC, 0.65; 95%CI: 0.53, 0.76; p = 0.001). For reader 2, the diagnostic performance of hrMRI (AUC, 0.97; 95%CI: 0.89, 1.00) was higher than that of cMRI (AUC, 0.74; 95%CI: 0.63, 0.84; p = 0.001), dMRI (AUC, 0.68; 95%CI: 0.56, 0.79; p = 0.001), and combined cMRI and dMRI (AUC, 0.70; 95%CI: 0.58, 0.80; p = 0.003).
Table 2 Diagnostic performance of cMRI, dMRI, and hrMRI for identifying pituitary microadenomas For reader 1, 23 of the 69 patients (33%) were misdiagnosed on both cMRI and dMRI, but 18 of the 23 misdiagnosed patients (78%) were correctly diagnosed on hrMRI. For reader 2, 17 of the 69 patients (25%) were misdiagnosed on both cMRI and dMRI, but 14 of the 17 misdiagnosed patients (82%) were correctly diagnosed on hrMRI.
Figure 2 shows that a 5-mm pituitary microadenoma was identified on preoperative pituitary MRI. The margin of the lesion was fully delineated on hrMRI, but not on cMRI and dMRI. Figure 3 shows that a 3-mm pituitary microadenoma was missed on cMRI, but identified on dMRI and hrMRI. Figure 4 shows that a 5-mm pituitary microadenoma was correctly diagnosed on hrMRI, but missed on cMRI or dMRI. Figure 5 shows that a 4-mm pituitary microadenoma was evident on coronal images as well as reconstructed axial and reconstructed sagittal images on hrMRI.
Fig. 2 
Images in a 56-year-old man with Cushing’s disease. The 5-mm pituitary microadenoma (arrow) can be identified on (a) coronal contrast-enhanced T1-weighted image and (b) coronal dynamic contrast-enhanced T1-weighted image obtained with two-dimensional (2D) fast spin echo (FSE) sequence, but the margin is not fully delineated. The lesion (arrow) is well delineated on (c) coronal contrast-enhanced T1-weighted image on high-resolution MRI obtained with 3D FSE sequence. d Intraoperative endoscopic photograph during transsphenoidal surgery after exposure of the sellar floor shows a round pituitary microadenoma (arrow)
Fig. 3 
Images in a 34-year-old woman with Cushing’s disease. No tumor is identified on (a) coronal contrast-enhanced T1-weighted image obtained with two-dimensional (2D) fast spin echo (FSE) sequence. The 3-mm pituitary microadenoma (arrow) with delayed enhancement is identified on the left side of the pituitary gland on (b) coronal dynamic contrast-enhanced T1-weighted image obtained with 2D FSE sequence and (c) coronal contrast-enhanced T1-weighted image on high-resolution MRI obtained with 3D FSE sequence. d Intraoperative endoscopic photograph during transsphenoidal surgery shows a 3-mm pituitary microadenoma (arrow)
Fig. 4 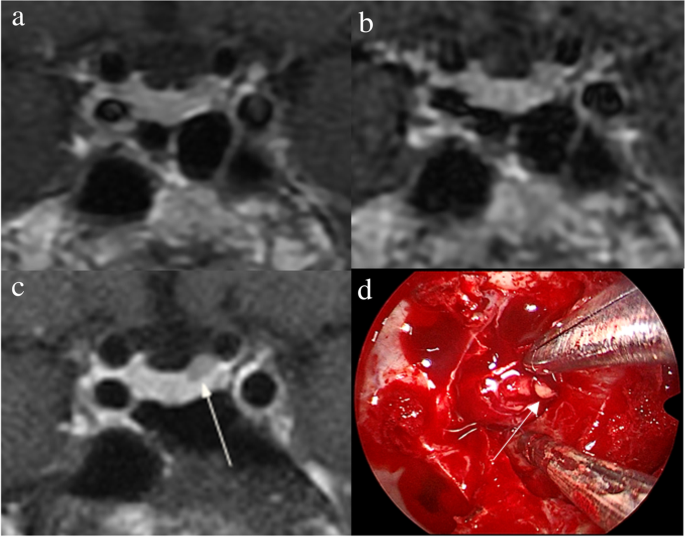
Images in a 43-year-old man with Cushing’s disease. The lesion is missed on (a) coronal contrast-enhanced T1-weighted image and (b) coronal dynamic contrast-enhanced T1-weighted image obtained with two-dimensional (2D) fast spin echo (FSE) sequence. c Coronal contrast-enhanced T1-weighted image on high-resolution MRI obtained with 3D FSE sequence shows a round pituitary microadenoma (arrow) measuring approximately 5 mm with delayed enhancement on the left side of the pituitary gland. d Intraoperative endoscopic photograph for microsurgical resection of the 5-mm pituitary microadenoma (arrow)
Fig. 5 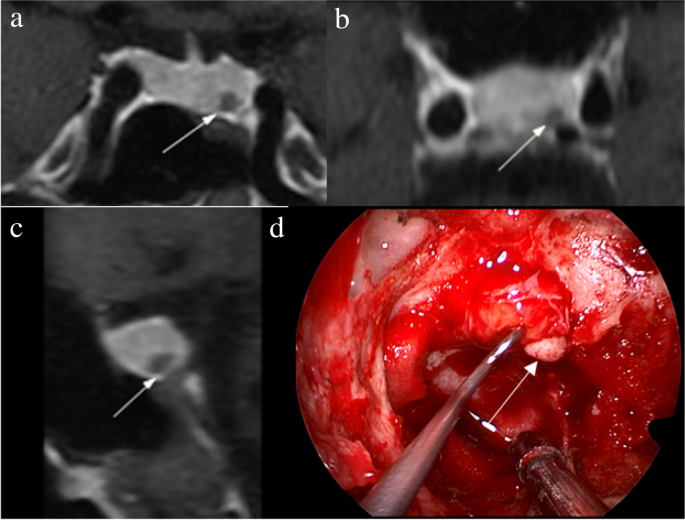
Images in a 48-year-old woman with Cushing’s disease. Preoperative high-resolution contrast-enhanced MRI using three-dimensional fast spin echo sequence shows a 4-mm pituitary microadenoma (arrow) with delayed enhancement is well delineated on the left side of the pituitary gland on (a) coronal, (b) reconstructed axial, and (c) reconstructed sagittal contrast-enhanced T1-weighted images. d Intraoperative endoscopic photograph during transsphenoidal surgery after exposure of the sellar floor shows a round pituitary microadenoma (arrow)
Image quality of cMRI, dMRI, and hrMRI
Image quality scores of cMRI, dMRI, and hrMRI are presented in Table 3. Scores for overall image quality, sharpness, and structural conspicuity on hrMRI (overall image quality, 5.0 [IQR, 5.0–5.0]; sharpness, 5.0 [IQR, 4.5–5.0]; structural conspicuity, 5.0 [IQR, 5.0–5.0]) were higher than those on cMRI (overall image quality, 4.0 [IQR, 3.5–4.0]; sharpness, 4.0 [IQR, 3.0–4.0]; structural conspicuity, 4.0 [IQR, 4.0–4.0]; p < 0.001 for all) and dMRI (overall image quality, 4.0 [IQR, 4.0–4.0]; sharpness, 4.0 [IQR, 4.0–4.0]; structural conspicuity, 4.0 [IQR, 4.0–4.5]; p < 0.001 for all).
Table 3 Image quality scores on cMRI, dMRI, and hrMRI The SNR and CNR measurements are shown in Table 4. The SNR of the pituitary microadenomas on hrMRI (67.5 [IQR, 51.2–92.1]) was lower than that on cMRI (82.3 [IQR, 61.8–127.2], p < 0.001), but higher than that on dMRI (53.9 [IQR, 35.2–72.6], p = 0.001). The CNR on hrMRI (26.2 [IQR, 15.1–41.0]) was higher than that on cMRI (10.6 [IQR, 0–42.6], p = 0.023) and dMRI (11.2 [IQR, 0–29.8], p < 0.001).
Table 4 SNR and CNR on cMRI, dMRI, and hrMRI Discussion
The identification of pituitary microadenomas is considerably challenging but critical in patients with ACTH-dependent Cushing’s syndrome. Our study demonstrated that hrMRI with 3D FSE sequence had higher diagnostic performance (AUC, 0.95–0.97) than cMRI (AUC, 0.74–0.75; p ≤ 0.002) and dMRI (AUC, 0.59–0.68; p ≤ 0.001) for identifying pituitary microadenomas. To our knowledge, there are no previous studies specifically evaluating the identification of pituitary microadenomas on hrMRI with 3D FSE sequence by comparison with cMRI and dMRI in patients with ACTH-dependent Cushing’s syndrome, and this is the largest study conducted in ACTH-secreting microadenomas with a sensitivity of more than 90%.
Recently, techniques for pituitary evaluation have developed rapidly. Because of false negatives and false positives on cMRI and dMRI using 2D FSE sequence [7, 9, 10], a 3D SPGR sequence was introduced for identifying pituitary adenomas. Previous studies demonstrated that the 3D SPGR sequence performed better than the 2D FSE sequence in the identification of pituitary adenomas with a sensitivity of up to 80% [11,12,13]. In patients with hyperprolactinemia, the 3D FSE sequence was recommended [14] and the 3D FSE sequence has rapidly developed recently with superior image quality [15, 16], suggesting that the 3D FSE sequence may be a reliable alternative for identifying pituitary adenomas. However, to our knowledge, few studies have investigated the diagnostic performance of the 3D FSE sequence for identifying ACTH-secreting pituitary adenomas. To fill the gaps, we conducted the current study and revealed that images obtained with the 3D FSE sequence had higher sensitivity (90–93%) in identifying pituitary microadenomas, than that in previous studies using the 3D SPGR sequence [8, 11,12,13].
There is a trade-off between spatial resolution and image noise. The reduced slice thickness can overcome the partial volume averaging effect, but it is associated with increased image noise [17]. Strikingly, our study showed that hrMRI had higher image quality scores than cMRI and dMRI, in terms of overall image quality, sharpness, and structural conspicuity. The SNR of the pituitary microadenomas on cMRI was slightly higher than that on hrMRI in our study. This is because the SNR was calculated as the mean signal intensity of the pituitary gland (instead of the pituitary microadenoma) divided by noise when no microadenoma was identified, and the mean signal intensity of the pituitary gland is higher than that of the pituitary microadenoma. About 40% of pituitary microadenomas were missed on cMRI, whereas less than 10% of pituitary microadenomas were missed on hrMRI. Given the situation mentioned above, the SNR on hrMRI was lower than that on cMRI. However, the CNR on hrMRI was significantly higher than that on cMRI and dMRI. Therefore, hrMRI in our study can dramatically improve the spatial resolution with high CNR, enabling the better identification of pituitary microadenomas.
The identification of pituitary adenomas on preoperative MRI in patients with ACTH-dependent Cushing’s syndrome could help the differential diagnosis of Cushing’s syndrome and aids surgical resection of lesions. It should be noted that most of the pituitary adenomas in patients with Cushing’s disease are microadenomas [5, 6]. In our study, all the tumors are microadenomas with a median diameter of 5 mm (IQR, 4–5 mm), making the diagnosis more challenging. The sensitivity of identifying pituitary adenomas decreased from 80 to 72% after excluding macroadenomas in a previous study [12], whereas the sensitivity of identifying pituitary microadenomas in our study was 90–93% on hrMRI. In the current study, hrMRI performed better than cMRI, dMRI, and combined cMRI and dMRI, with high AUC (0.95–0.97), high sensitivity (90–93%), and high specificity (100%), superior to previous studies [8, 11,12,13]. The high sensitivity of hrMRI for identifying pituitary adenomas will help surgeons improve the postoperative remission rate [4]. The high specificity of hrMRI will assist clinicians to consider ectopic ACTH syndrome, and then perform imaging to identify ectopic tumors. Besides, the inter-observer agreement for identifying pituitary microadenomas was almost perfect on hrMRI (κ = 0.91), which was moderate on cMRI (κ = 0.50) and dMRI (κ = 0.57). Therefore, hrMRI using the 3D FSE sequence is a potential alternative that can significantly improve the identification of pituitary microadenomas.
Limitations of the study included its retrospective nature and the relatively small sample size in patients with ectopic ACTH syndrome as negative controls. The bias may be introduced in the patient inclusion process. Only those patients who underwent all the cMRI, dMRI, and hrMRI scans were included. In fact, some patients will bypass hrMRI when obvious pituitary adenomas were detected on cMRI and dMRI. These patients were not included in the current study because of lack of hrMRI findings. Given the situation, the sensitivity of identifying pituitary adenomas will be higher with the enrollment of these patients. Besides, the timing of the sequence acquisition after contrast injection is essential [16] and bias may be introduced due to the postcontrast enhancement curve of both the pituitary gland and the microadenoma [14]. In the future, a prospective study with different sequence acquisition orders is needed to minimize possible interference caused by the postcontrast enhancement curve. Moreover, a larger sample size is also needed to verify the diagnostic performance of hrMRI using 3D FSE sequence for identifying pituitary microadenomas and to determine whether it can replace 2D FSE or 3D SPGR sequences for routinely evaluating the pituitary gland.
In conclusion, hrMRI with 3D FSE sequence showed higher diagnostic performance than cMRI and dMRI for identifying pituitary microadenomas in patients with Cushing’s syndrome.
Abbreviations
- ACTH:
-
Adrenocorticotropic hormone
- AUC:
-
Area under the receiver operating characteristics curve
- cMRI:
-
Conventional contrast-enhanced MRI
- CNR:
-
Contrast-to-noise ratio
- dMRI:
-
Dynamic contrast-enhanced MRI
- FSE:
-
Fast spin echo
- hrMRI:
-
High-resolution contrast-enhanced MRI
- IQR:
-
Interquartile range
- SNR:
-
Signal-to-noise ratio
- SPGR:
-
Spoiled gradient re
-
called
References
-
Lacroix A, Feelders RA, Stratakis CA, Nieman LK (2015) Cushing’s syndrome. Lancet 386:913–927
-
Loriaux DL (2017) Diagnosis and differential diagnosis of Cushing’s syndrome. N Engl J Med 376:1451–1459
-
Nieman LK, Biller BM, Findling JW et al (2015) Treatment of Cushing’s syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 100:2807–2831
-
Yamada S, Fukuhara N, Nishioka H et al (2012) Surgical management and outcomes in patients with Cushing disease with negative pituitary magnetic resonance imaging. World Neurosurg 77:525–532
-
Vitale G, Tortora F, Baldelli R et al (2017) Pituitary magnetic resonance imaging in Cushing’s disease. Endocrine 55:691–696
-
Jagannathan J, Smith R, DeVroom HL et al (2009) Outcome of using the histological pseudocapsule as a surgical capsule in Cushing disease. J Neurosurg 111:531–539
-
Boscaro M, Arnaldi G (2009) Approach to the patient with possible Cushing’s syndrome. J Clin Endocrinol Metab 94:3121–3131
-
Kasaliwal R, Sankhe SS, Lila AR et al (2013) Volume interpolated 3D-spoiled gradient echo sequence is better than dynamic contrast spin echo sequence for MRI detection of corticotropin secreting pituitary microadenomas. Clin Endocrinol (Oxf) 78:825–830
-
Lonser RR, Nieman L, Oldfield EH (2017) Cushing’s disease: pathobiology, diagnosis, and management. J Neurosurg 126:404–417
-
Potts MB, Shah JK, Molinaro AM et al (2014) Cavernous and inferior petrosal sinus sampling and dynamic magnetic resonance imaging in the preoperative evaluation of Cushing’s disease. J Neurooncol 116:593–600
-
Grober Y, Grober H, Wintermark M, Jane JA, Oldfield EH (2018) Comparison of MRI techniques for detecting microadenomas in Cushing’s disease. J Neurosurg 128:1051–1057
-
Fukuhara N, Inoshita N, Yamaguchi-Okada M et al (2019) Outcomes of three-Tesla magnetic resonance imaging for the identification of pituitary adenoma in patients with Cushing’s disease. Endocr J 66:259–264
-
Patronas N, Bulakbasi N, Stratakis CA et al (2003) Spoiled gradient recalled acquisition in the steady state technique is superior to conventional postcontrast spin echo technique for magnetic resonance imaging detection of adrenocorticotropin-secreting pituitary tumors. J Clin Endocrinol Metab 88:1565–1569
-
Magnaldi S, Frezza F, Longo R, Ukmar M, Razavi IS, Pozzi-Mucelli RS (1997) Assessment of pituitary microadenomas: comparison between 2D and 3D MR sequences. Magn Reson Imaging 15:21–27
-
Lien RJ, Corcuera-Solano I, Pawha PS, Naidich TP, Tanenbaum LN (2015) Three-Tesla imaging of the pituitary and parasellar region: T1-weighted 3-dimensional fast spin echo cube outperforms conventional 2-dimensional magnetic resonance imaging. J Comput Assist Tomogr 39:329–333
-
Sartoretti T, Sartoretti E, Wyss M et al (2019) Compressed SENSE accelerated 3D T1w black blood turbo spin echo versus 2D T1w turbo spin echo sequence in pituitary magnetic resonance imaging. Eur J Radiol 120:108667
-
Kim M, Kim HS, Kim HJ et al (2021) Thin-slice pituitary MRI with deep learning-based reconstruction: diagnostic performance in a postoperative setting. Radiology 298:114–122
Acknowledgements
We thank Dr. Kai Sun, Medical Research Center, Peking Union Medical College Hospital, for his guidance on the statistical analysis in this study.
Funding
This study has received funding from the National Natural Science Foundation of China (grant 82071899), the National Key Research and Development Program of China (grants 2016YFC1305901, 2020YFA0804500), the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (grants 2017-I2M-3–008, 2021-I2M-1–025), the Beijing Natural Science Foundation (grant L182067) and National High Level Hospital Clinical Research Funding (2022-PUMCH-B-067, 2022-PUMCH-B-114).
Ethics declarations
Guarantor
The scientific guarantor of this publication is Feng Feng.
Conflict of interest
The authors of this manuscript declare no conflict of interest.
Statistics and biometry
No complex statistical methods were necessary for this paper.
Informed consent
Written informed consent was waived by the Institutional Review Board.
Ethical approval
Institutional Review Board approval was obtained.
Methodology
• retrospective
• diagnostic or prognostic study
• performed at one institution
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
From https://link.springer.com/article/10.1007/s00330-023-09585-1
-
-
1
-
From Janice: A devastating horrific disease that doctors won’t believe we have. Feeling like we will collapse with each step. Carrying an empty plastic bag seems too heavy to manage. And no one will believe us. They think we are lazy, fat and crazy.
What is your experience?
-
1
-
-
Abstract
Importance Cushing syndrome is defined as a prolonged increase in plasma cortisol levels that is not due to a physiological etiology. Although the most frequent cause of Cushing syndrome is exogenous steroid use, the estimated incidence of Cushing syndrome due to endogenous overproduction of cortisol ranges from 2 to 8 per million people annually. Cushing syndrome is associated with hyperglycemia, protein catabolism, immunosuppression, hypertension, weight gain, neurocognitive changes, and mood disorders.
Observations Cushing syndrome characteristically presents with skin changes such as facial plethora, easy bruising, and purple striae and with metabolic manifestations such as hyperglycemia, hypertension, and excess fat deposition in the face, back of the neck, and visceral organs. Cushing disease, in which corticotropin excess is produced by a benign pituitary tumor, occurs in approximately 60% to 70% of patients with Cushing syndrome due to endogenous cortisol production. Evaluation of patients with possible Cushing syndrome begins with ruling out exogenous steroid use. Screening for elevated cortisol is performed with a 24-hour urinary free cortisol test or late-night salivary cortisol test or by evaluating whether cortisol is suppressed the morning after an evening dexamethasone dose. Plasma corticotropin levels can help distinguish between adrenal causes of hypercortisolism (suppressed corticotropin) and corticotropin-dependent forms of hypercortisolism (midnormal to elevated corticotropin levels). Pituitary magnetic resonance imaging, bilateral inferior petrosal sinus sampling, and adrenal or whole-body imaging can help identify tumor sources of hypercortisolism. Management of Cushing syndrome begins with surgery to remove the source of excess endogenous cortisol production followed by medication that includes adrenal steroidogenesis inhibitors, pituitary-targeted drugs, or glucocorticoid receptor blockers. For patients not responsive to surgery and medication, radiation therapy and bilateral adrenalectomy may be appropriate.
Conclusions and Relevance The incidence of Cushing syndrome due to endogenous overproduction of cortisol is 2 to 8 people per million annually. First-line therapy for Cushing syndrome due to endogenous overproduction of cortisol is surgery to remove the causative tumor. Many patients will require additional treatment with medications, radiation, or bilateral adrenalectomy.
-
1
-
-
In this application note, Tecan presents a method for diagnosing Cushing's syndrome efficiently and accurately. The approach involves simultaneous the measurement of cortisol and dexamethasone levels using LC-MS/MS, which reduces false positives in dexamethasone suppression test (DSTs). The described LC-MS/MS method enables the tracking of multiple analytes, including cortisol, cortisone, and dexamethasone, in serum or plasma. Implementing this analytical approach offers clinical laboratories a straightforward means of performing DSTs, and the availability of a commercially available kit ensures reliable and reproducible results.
-
1
-
-
Abstract
Rationale:
Ectopic ACTH-producing pituitary adenoma (EAPA) of the clivus region is extraordinarily infrequent condition and merely a few reports have been reported to date.
Patient concerns:
The patient was a 53-year-old woman who presented with Cushing-like appearances and a soft tissue mass in the clivus region.
Diagnoses:
The final diagnosis of clivus region EAPA was established by clinical, radiological and histopathological findings.
Interventions:
The patient underwent gross total clivus tumor resection via transsphenoidal endoscopy.
Outcomes:
Half a year after surgery, the patient Cushing-like clinical manifestations improved significantly, and urinary free cortisol and serum adrenocorticotropin (ACTH) returned to normal.
Lessons:
Given the extreme scarcity of these tumors and their unique clinical presentations, it may be possible to misdiagnose and delayed treatment. Accordingly, it is especially crucial to summarize such lesions through our present case and review the literature for their precise diagnosis and the selection of optimal treatment strategies.
1. Introduction
Pituitary adenoma arises from the anterior pituitary cells and is the commonest tumor of the sellar region.[1] It makes up approximately 10% to 15% of all intracranial tumors.[2] Ectopic pituitary adenoma (EPA) is defined as a pituitary adenoma that occurs outside the sellar area and has no direct connection to normal pituitary tissue.[3] The most frequent sites of EPA are the sphenoid sinus and suprasellar region, and much less frequent sites including the clivus region, cavernous sinus, and nasopharynx.[4]
Hypercortisolism and the series of symptoms it leads to is termed Cushing syndrome (CS).[5] CS is classified into adrenocorticotropin (ACTH)-dependent and ACTH-independent CS depending on the cause, accounting for 80% to 85% and 15% to 20% of cases, respectively.[6] Pituitary adenoma accounts for ACTH-dependent CS 75% to 80%, while ectopic ACTH secretion accounts for the remaining 15% to 20%.[7] Ectopic CS is a very rare disorder of CS caused by an ACTH-secreting tumor outside the pituitary or adrenal gland.[8] It has been reported that ectopic ACTH-producing pituitary adenoma (EAPA) can occur in the sphenoid sinus, cavernous sinus, clivus, and suprasellar region,[9] with EAPA in the clivus region being extremely rare, and merely 6 cases have been reported in the English literature (Table 1).[10–15] Furthermore, as summarized in the Table 1, EAPA in the clivus area has unique symptoms, which may lead to misdiagnosis as well as delay in treatment. Therefore, we herein described a case of CS from an EAPA of the clivus region and reviewed relevant literature for the purpose of further understanding this extraordinarily unusual condition.
Table 1 - Literature review of cases of primary clival ectopic ACTH-producing pituitary adenoma (including the current case).Reference Age (yr)/sex Symptoms Imaging findings Maximum tumor diameter (mm) Preoperative elevated hormone IHC Surgery RT Follow-up (mo) Outcome Ortiz et al 1975[10] 15/F NA NA NA NA NA Right transfrontal craniotomy, NA Yes NA Symptomatic relief Anand et al 1993[11] 58/F Anosphrasia, blurred vision, occasional left frontal headache, Routine radiographic evaluation revealed a clival tumor and nasopharyngeal mass with bone erosion. MRI demonstrated a Midline homogeneous mass. 30 ACTH ACTH in a few isolated cells Maxillotomy approach, GTR Yes 12 Symptomatic relief Pluta et al 1999[12] 20/F Cushing syndrome MRI revealed a hypodense contrast-enhancing lesion. NA ACTH ACTH Transsphenoidal surgery, GTR No 18 Symptomatic relief Shah et al 2011[13] 64/M Facial paresthesias, myalgias, decreased muscle strength, and fatigue CT imaging showed a clival mass. 21 ACTH ACTH NA, GTR No 7 Symptomatic relief Aftab et al 2021[14] 62/F Transient unilateral visual loss MRI showed a T2 heterogeneously enhancing hyperintense lesion. 21 No ACTH Transsphenoidal resection, GTR NO 6 Symptomatic relief Li et al 2023[15] 47/F Bloody nasal discharge, dizziness and headache CT revealed an ill-defined mass eroding the adjacent bone. MRI T1 showed a heterogeneous mass with hypointensity, hyperintensity on T2-weighted images and isointensity on diffusion-weighted images. 58 NA ACTH Transsphenoidal endoscopy, STR Yes 2 Symptomatic relief Current case 53/F Headache, and dizziness, Cushing syndrome CT demonstrated bone destruction and a soft tissue mass. MRI T1 revealed irregular isointense signal, and MRI T2 showed isointense signal/slightly high signal. 46 ACTH ACTH Transsphenoidal endoscopy, GTR NO 6 Symptomatic relief ACTH = adrenocorticotropin, CT = computed tomography, GTR = gross total resection, IHC = immunohistochemistry, MRI = magnetic resonance imaging, NA = not available, RT = radiotherapy, STR = subtotal resection.2. Case presentation
A 53-year-old female presented to endocrinology clinic of our hospital with headache and dizziness for 2 years and aggravated for 1 week. Her past medical history was hypertension, with blood pressure as high as 180/100 mm Hg. Her antihypertensive medications included amlodipine besylate, benazepril hydrochloride, and metoprolol tartrate, and she felt her blood pressure was well controlled. In addition, she suffered a fracture of the thoracic vertebrae 3 month ago; and bilateral rib fractures 1 month ago. Physical examination revealed that the patient presented classical Cushing-like appearances, including moon face and supraclavicular and back fat pads, and centripetal obesity (body mass index, 25.54 kg/m2) with hypertension (blood pressure, 160/85 mm Hg).
Laboratory studies revealed high urinary free cortisol levels at 962.16 µg/24 hours (reference range, 50–437 µg/24 hours) and absence of circadian cortisol rhythm (F [0am] 33.14 µg/dL, F [8am] 33.52 µg/dL, F [4pm] 33.3 µg/dL). ACTH levels were elevated at 90.8 pg/mL (reference range, <46 pg/mL). The patient low-dose dexamethasone suppression test demonstrated the existence of endogenous hypercortisolism. High-dose dexamethasone suppression test results revealed that serum cortisol levels were suppressed by <50%, suggesting the possibility of ectopic ACTH-dependent CS. Serum luteinizing hormone and serum follicle stimulating hormone were at low levels, <0.07 IU/L (reference range, 15.9–54.0 IU/L) and 2.57 IU/L (reference range, 23.0–116.3 IU/L), respectively. Insulin-like growth factor-1, growth hormone (GH), prolactin (PRL), thyroid stimulating hormone, testosterone, progesterone and estradiol test results are all normal. Oral glucose tolerance test showed fasting glucose of 6.3 mmol/L and 2-hour glucose of 18.72 mmol/L; glycosylated hemoglobin (HbA1c) was 7.1%. Serum potassium fluctuated in the range of 3.14 to 3.38 mmol/L (reference range, 3.5–5.5 mmol/L), indicating mild hypokalemia.
High-resolution computed tomography (CT) scan of the sinuses revealed osteolytic bone destruction of the occipital clivus and a soft tissue mass measuring 20 mm × 30 mm × 46 mm (Fig. 1A). The mass filled the bilateral sphenoid sinuses and involved the cavernous sinuses, but the pituitary was normal. Cranial MR scan showed the T1W1 isointense signal and the T2W1 isointense signal/slightly high signal in the sphenoid sinus and saddle area (Fig. 1B–D). Bone density test indicated osteoporosis.

Figure 1.: Radiological findings. (A) CT demonstrated bone destruction and a soft tissue mass on the occipital clivus (white arrow). (B) Axial view of the MR T1 revealed irregular isointense signal in the sphenoid sinus and saddle area (white arrow). (C and D) Axial view and sagittal view of the MR T2 showed isointense signal/slightly high signal in the sphenoid sinus and saddle area (black arrow). CT = computed tomography.Subsequently, the patient underwent gross total clivus tumor resection via transsphenoidal endoscopy. During surgery, the tumor was found to be light red in color with a medium texture, and the tumor tissue protruded into the sphenoidal sinus cavity and eroded the clival area. Histologically, the tumor cells were nested, with interstitially rich blood sinuses and organoid arrangement (Fig. 2A). The tumor cells were relatively uniform in size, with light red cytoplasm, delicate pepper salt-like chromatin, and visible nucleoli (Fig. 2B). In addition, mitosis of tumor cells was extremely rare. Immunohistochemically, the neoplasm cells were diffuse positive for CK (Fig. 2C), CgA (Fig. 2D), ACTH (Fig. 2E), Syn and CAM5.2, with low Ki-67 labeling index (<1%) (Fig. 2F). Simultaneously, all other pituitary hormone markers like GH, thyroid stimulating hormone, PRL, luteinizing hormone, as well as follicle stimulating hormone were negatively expressed. On the basis of these medically historical, clinical, laboratorial, morphologic, and immunohistochemical findings, the final pathological diagnosis of an EAPA was established.

Figure 2.: HE and immunohistochemical findings. (A) Histologic sections revealed morphologically homogeneous tumor cells in nests with a prominent and delicate vascularized stroma (H&E, × 200). (B) The tumor cells had fine chromatin with visible nuclei and rare mitoses (H&E, × 400). CK (C), CgA (D) and ACTH (E) immunohistochemically showed diffuse reactivity of the tumor cells (SP × 200). (F) The proliferation index is <1% on Ki-67 staining (SP × 200).When evaluated 2 months after surgery, her Cushing-like characteristics had well improved, and her blood pressure was normal. Furthermore, her serum cortisol and ACTH returned to the normal levels. Six-month postoperative follow-up revealed that serum cortisol and ACTH were stable at normal levels, and no signs of tumor recurrence were detected on imaging.
3. Discussion
EAPA is defined as an ACTH-secreting ectopic adenoma located outside the ventricles, and has no continuity with the normal intrasellar pituitary gland.[9] ACTH promotes cortisol secretion by stimulating the adrenal cortical fasciculus. The clinical manifestations of hypercortisolism are diverse, and the severity is partly related to the duration of the cortisol increase.[8] Clival tumors are typically uncommon, accounting for 1% of all intracranial tumors. There are many differential diagnoses for clival lesions, including the most common chordoma (40%), meningioma, chondrosarcoma, astrocytoma, craniopharyngioma, germ cell tumors, non-Hodgkin lymphoma, melanoma, metastatic carcinoma, and rarely pituitary adenoma.[16] The commonest clival EPA is a PRL adenoma, followed by null cell adenoma, and the least common are ACTH adenoma and GH adenoma.[2] The clival EAPA is extremely unwonted, and only 6 other cases apart from ours have been reported in literature so far (Table 1).
The average age of the patients with these tumors was 48 years (range, 15–64 years). There was a obvious female predominance with a female-to-male prevalence ratio of 6:1. Only 2 patients (2/6, 33.3%) with reported clinical symptoms, including our patients, presented with overt clinical manifestations of CS. Compression of the mass on adjacent structures (e.g., nerves) may result in anosphrasia, visual impairment, headache, myalgias, decreased muscle strength, dizziness and facial sensory abnormalities. The diagnosis and localization of these tumors relied heavily on radiological imaging. Head MRI was the most basic method used for them detection, for localization adenomas and their invasion of surrounding structures to guide the choice of treatment and surgical options methods. Radiographic characteristics had been reported in 6 patients with EAPA in the clivus region. All of these patients (6/6, 100%) had initial positive findings of sellar MRI (or CT) identifying an ectopic adenoma before surgery. MR T1 was usually a low-intensity or isointense signal, while MR T2 was usually an isointense or slightly higher signal. The maximum diameter of the tumor was reported in 5 cases, with the mean maximum diameter was 35.2 mm (range, 21–55 mm) according to preoperative MRI and intraoperative observations. As summarized in Table 1, 4/5 clival EAPA cases secreted ACTH. Histologically, all cases (6/6, 100%) expressed ACTH scatteredly or diffusely.
The gold standard for the treatment of CS caused by EAPA was the surgical removal of EPA, which was essential to achieve remission and histological confirmation of the disease.[9] The most common method of EAPA resection in the clivus region was transsphenoidal sinus resection (4/6, 66.67%), followed by craniotomy (1/6, 16.67%) and maxillary osteotomy (1/6, 16.67%). Transsphenoidal endoscopic surgery allowed resection of the EAPA and manipulation of neurovascular structures and avoidance of cerebral atrophy, whereas craniotomy allowed full exposure of the suprasellar region, direct visualization or manipulation of the adenoma, and reduced the risk of postoperative CSF leak.[9] Both approaches had their advantages, and there was no consensus on which surgical approach was best for the treatment of EAPA in the slope area.[9] The choice of the best surgical approach was believed to be based on the condition of the adenoma, as well as the general condition of the patient and the experience of the surgeon.[9] As summarized in Table 1, most complete tumor resections were achieved regardless of the method chosen. A minority of patients underwent postoperative radiotherapy (3/7, 42.86%), and most of them had invasion of the surrounding bone tissue. All patients experienced effective postoperative relief of symptoms.
In summary, due to the rarity of this disorder, an accurate preoperative diagnosis of EAPA in the slope area is extremely challenging for the clinician or radiologist. The final precise diagnosis relies on a combination of clinical symptoms, imaging findings, histology and immunohistochemical markers. For this type of tumor, surgery is an effective treatment to relieve the clinical manifestations caused by tumor compression or hormonal secretion. The choice of postoperative adjuvant radiotherapy is mainly based on the presence of invasion of the surrounding bone tissue. Further cases may be necessary to summarize the clinical features of such lesions and to develop optimal treatment strategies.
Acknowledgments
We would like to thank the patient and her family.
Author contributions
Conceptualization: Yutao He.
Data curation: Ziyi Tang.
Formal analysis: Na Tang.
Methodology: Yu Lu, Fangfang Niu, Jiao Ye, Zheng Zhang, Chenghong Fang.
Writing – original draft: Yutao He.
Writing – review & editing: Yutao He, Lei Yao.
Abbreviations:
- ACTH
- adrenocorticotropin
- CS
- cushing syndrome
- CT
- computed tomography
- EAPA
- ectopic ACTH-producing pituitary adenoma
- EPA
- ectopic pituitary adenoma
- GH
- growth hormone
- PRL
- prolactin
References
[1]. Gittleman H, Ostrom QT, Farah PD, et al. Descriptive epidemiology of pituitary tumors in the United States, 2004-2009. J Neurosurg. 2014;121:527–35.[2]. Karras CL, Abecassis IJ, Abecassis ZA, et al. Clival ectopic pituitary adenoma mimicking a Chordoma: case report and review of the literature. Case Rep Neurol Med. 2016;2016:8371697.[3]. Bălaşa AF, Chinezu R, Teleanu DM, et al. Ectopic intracavernous corticotroph microadenoma: case report of an extremely rare pathology. Rom J Morphol Embryol. 2017;58:1447–51.[4]. Zhu J, Wang Z, Zhang Y, et al. Ectopic pituitary adenomas: clinical features, diagnostic challenges and management. Pituitary. 2020;23:648–64.[5]. Paleń-Tytko JE, Przybylik-Mazurek EM, Rzepka EJ, et al. Ectopic ACTH syndrome of different origin-diagnostic approach and clinical outcome. experience of one clinical centre. PLoS One. 2020;15:e0242679.[6]. Sharma ST, Nieman LK, Feelders RA. Cushing’s syndrome: epidemiology and developments in disease management. Clin Epidemiol. 2015;7:281–93.[7]. Aniszewski JP, Young WF Jr, Thompson GB, et al. Cushing syndrome due to ectopic adrenocorticotropic hormone secretion. World J Surg. 2001;25:934–40.[8]. Mohib O, Papleux E, Remmelink M, et al. An ectopic Cushing’s syndrome as a cause of severe refractory hypokalemia in the ICU. Acta Clin Belg. 2021;76:373–8.[9]. Sun X, Lu L, Feng M, et al. Cushing syndrome caused by ectopic adrenocorticotropic hormone-secreting pituitary adenomas: case report and literature review. World Neurosurg. 2020;142:75–86.[10]. Ortiz-Suarez H, Erickson DL. Pituitary adenomas of adolescents. J Neurosurg. 1975;43:437–9.[11]. Anand VK, Osborne CM, Harkey HL. Infiltrative clival pituitary adenoma of ectopic origin. Otolaryngol Head Neck Surg. 1993;108:178–83.[12]. Pluta RM, Nieman L, Doppman JL, et al. Extrapituitary parasellar microadenoma in Cushing’s disease. J Clin Endocrinol Metab. 1999;84:2912–23.[13]. Shah R, Schniederjan M, DelGaudio JM, et al. Visual vignette.s Ectopic ACTH-secreting pituitary adenoma. Endocr Pract. 2011;17:966.[14]. Aftab HB, Gunay C, Dermesropian R, et al. “An Unexpected Pit” - ectopic pituitary adenoma. J Endocr Soc. 2021;5:A557–8.[15]. Li Y, Zhu JG, Li QQ, et al. Ectopic invasive ACTH-secreting pituitary adenoma mimicking chordoma: a case report and literature review. BMC Neurol. 2023;23:81.[16]. Wong K, Raisanen J, Taylor SL, et al. Pituitary adenoma as an unsuspected clival tumor. Am J Surg Pathol. 1995;19:900–3.Keywords:clivus region; Cushing; Ectopic ACTH; like appearance; producing pituitary adenoma
-
1
-
On July 6, 1953, the NIH Clinical Center opened its doors to patients. Oveta Culp Hobby, the Secretary of Welfare, said at the time, “We are now carrying on in the United States the most intensive and widespread research attack on human disease that the world has ever seen.”
Seventy years later, the Clinical Center remains a national focal point for clinical research and creating cures that improve the health of the nation and the world.
Upcoming Events
Check back for more details!Anniversary Celebration
July 2023
Lipsett AuditoriumScientist Artists Exhibit
Summer 2023
Clinical CenterIF/THEN Exhibit
Summer 2023
Clinical Center Arboretums70th anniversary Grand Rounds Lecture
June 28, 2023, noon – 1 pm
Lipsett AuditoriumLecture features former NIH Director Dr. Francis Collins. Limited onsite seating. Also available on NIH Videocast.
More info at https://www.cc.nih.gov/ocmr/history/70thanniversary.html
-
In Italy it is estimated that there are about 3,000 patients suffering from Cushing’s syndrome, while in Europe the number rises to over 50,000.
The Cushing’s syndrome, a disease caused by the excessive production of cortisol by the pituitary gland due to a benign tumor of the gland, has seen a breakthrough in its treatment. Thanks to a new drug called osilodrostat, approved in 2020 by the Food and Drug Administration and subsequently by Aifa in Italy, patients unfit for surgery can benefit from a treatment that offers the same effects as a scalpel. Furthermore, this drug reduced symptoms in 80% of cases.
Cushing’s syndrome has been dubbed “full moon face disease” due to its most obvious visible effects, such as a rounding of the face caused by fat accumulation and visible weight gain also on the waist and back. Despite its symptomatic relevance, the disease has long been poorly understood by both healthcare professionals and the general public. To raise awareness of this syndrome, the #Thiscushing campaign has been launched, which aims to spread knowledge about the disease. The campaign recently stopped in Rome, during the Congress of the Italian Society of Endocrinology (SIE), where a photographic exhibition was organized which represents moments of daily life of people affected by Cushing’s syndrome and their difficulties.
Despite the debilitating symptoms, Cushing’s syndrome is often underdiagnosed, resulting in delays in diagnosis of up to 5-7 years. The disease presents a wide range of symptoms, ranging from difficulty performing even simple daily activities such as tying your shoes or getting out of bed, to common manifestations such as high cholesterol, hypertension and hyperglycemia, which can be confused with symptoms of other less common pathologies. serious. It is for this reason that the EIS experts are appealing for the inclusion of Cushing’s syndrome in the list of rare pathologies recognized by the Ministry of Health, in order to facilitate timely diagnosis and faster access to the necessary treatments.
-
1
-
-

YOU’RE INVITED! GoodHormoneHealth Webinar on Oh-Oh-Oh-Ozempic
Dr. Theodore Friedman (The Wiz) will giving a webinar on Ozempic and other new weight loss medicines.Topics to be discussed include:
- Who should go on weight-loss medications?
- Which weight-loss medications are the best?
- What are the side effects?
- How do they work with diet and exercise?
- How do you get insurance coverage?
- There will be an opportunity for patients to share their experience on Facebook
Sunday • Jul 9, 2023 • 6 PM PDTVia Zoom Click here to join the meeting or
https://us02web.zoom.us/j/4209687343?pwd= amw4UzJLRDhBRXk1cS9ITU02V1pEQT 09
OR
+16699006833,,4209687343#,,,,*111116#
OR
Join on Facebook Live - https://www.facebook.com/goodhormonehealth
Slides will be available on the day of the talk here.
There will be plenty of time for questions using the chat button.
For more information, email us at mail@goodhormonehealth.com-
1
-
Abstract
Background
The diagnosis of Cushing’s syndrome is challenging; however, through the clinical picture and the search for secondary causes of osteoporosis, it was possible to reach the diagnosis of the case reported. There was an independent, symptomatic ACTH hypercortisolism manifested by typical phenotypic changes, severe secondary osteoporosis and arterial hypertension in a young patient.
Case presentation
A 20-year-old Brazilian man with low back pain for 8 months. Radiographs showed fragility fractures in the thoracolumbar spine, and bone densitometry showed osteoporosis, especially when evaluating the Z Score (− 5.6 in the lumbar spine). On physical examination, there were wide violaceous streaks on the upper limbs and abdomen, plethora and fat increase in the temporal facial region, hump, ecchymosis on limbs, hypotrophy of arms and thighs, central obesity and kyphoscoliosis. His blood pressure was 150 × 90 mmHg. Cortisol after 1 mg of dexamethasone (24.1 µg/dL) and after Liddle 1 (28 µg/dL) were not suppressed, despite normal cortisoluria. Tomography showed bilateral adrenal nodules with more severe characteristics. Unfortunately, through the catheterization of adrenal veins, it was not possible to differentiate the nodules due to the achievement of cortisol levels that exceeded the upper limit of the dilution method. Among the hypotheses for the differential diagnosis of bilateral adrenal hyperplasia are primary bilateral macronodular adrenal hyperplasia, McCune–Albright syndrome and isolated bilateral primary pigmented nodular hyperplasia or associated with Carney’s complex. In this case, primary pigmented nodular hyperplasia or carcinoma became important etiological hypotheses when comparing the epidemiology in a young man and the clinical-laboratory-imaging findings of the differential diagnoses. After 6 months of drug inhibition of steroidogenesis, blood pressure control and anti-osteoporotic therapy, the levels and deleterious metabolic effects of hypercortisolism, which could also impair adrenalectomy in the short and long term, were reduced. Left adrenalectomy was chosen, given the possibility of malignancy in a young patient and to avoid unnecessary definitive surgical adrenal insufficiency if the adrenalectomy was bilateral. Anatomopathology of the left gland revealed expansion of the zona fasciculate with multiple nonencapsulated nodules.
Conclusion
The early identification of Cushing’s syndrome, with measures based on the assessment of risks and benefits, remains the best way to prevent its progression and reduce the morbidity of the condition. Despite the unavailability of genetic analysis for a precise etiological definition, it is possible to take efficient measures to avoid future damage.
Background
Cushing’s syndrome may be exogenous or endogenous and, in this case, can be ACTH-dependent or independent. In the case reported, there was an independent, symptomatic ACTH hypercortisolism manifested by typical phenotypic changes, severe secondary osteoporosis and arterial hypertension in a young patient. Osteoporosis secondary to hypercortisolism occurs due to chronic reduction in bone formation, loss of osteocytes and increased reabsorption caused by intense binding of cortisol to glucocorticoid receptors present in bone cells [1]. In addition, excess cortisol impairs vitamin D metabolism and reduces endogenous parathyroid hormone secretion, intestinal calcium reabsorption, growth hormone release, and lean body mass [2]. Subclinical Cushing disease occurs in up to 11% of individuals diagnosed with early-onset osteoporosis and 0.5–1% of hypertension patients. [3] A cross-sectional study published in 2023 revealed a prevalence of 81.5% bone loss in 19 patients with Cushing’s syndrome [2]. The prevalence of osteopenia ranges from 60 to 80%, and the prevalence of osteoporosis ranges from 30 to 65% in patients with Cushing’s syndrome. Additionally, the incidence of fragility fractures ranges from 30 to 50% in these patients [4] and is considered the main cause of morbidity affecting the quality of life. The diagnosis is challenging, given the presence of confounding factors; however, through the clinical picture and the search for secondary causes of osteoporosis, it was possible to reach a syndromic diagnosis. Early identification of this syndrome, with measures based on the assessment of risks and benefits, remains the best way to prevent progression and reduce morbidity related to this disease [2].
Case presentation
A 20-year-old Brazilian male patient reported low back pain that had evolved for 8 months, with no related trauma. He sought emergency care and performed spinal radiographs on this occasion (03/2019). Due to the several alterations observed in the images, he was referred to the Orthopedics Service of the Hospital of Federal University of Juiz de Fora, which prescribed orthopedic braces, indicated physical therapy and was referred again to the Osteometabolic Diseases outpatient clinic of the Endocrinology and Rheumatology Services of the Hospital of Federal University of Juiz de Fora on 10/2019.
The radiographs showed a marked reduction in the density of bone structures, scoliotic deviation with convexity toward the left and reduction in the height of the lumbar vertebrae, with partial collapses of the vertebral bodies at the level of T12, L1, L2, L3 and L5, with recent collapses in T12 and L1, suggesting bone fragility fractures. The same can be seen in posterior magnetic resonance imaging (Fig. 1).
Fig. 1 
Radiography and Magnetic Resonance Imaging (MRI) of lumbosacral spine in profile
Bone scintigraphy on 08/2019 did not reveal hyper flow or anomalous hyperemia in the topography of the thoracolumbar spine, and in the later images of the exam, there was a greater relative uptake of the tracer in the lumbar spine (vertebrae T10–T12, L2–L4), of nonspecific aspect, questioning the presence of osteoarticular processes or ankylosing spondylitis.
It was also observed in the bone densitometry requested in October 2019, performed by dual-energy X-ray absorptiometry (DXA), low bone mineral density (BMD) in the lumbar spine, femoral neck and total femur, when comparing the results to evaluating the Z Score (Table 1).
Table 1 Dual-energy X-ray absorptiometry (DXA) Thus, the diagnosis of osteoporosis was established, and treatment with vitamin D 7000 IU per week was started due to vitamin D3 insufficiency associated with the bisphosphonate alendronate 70 mg, also weekly. The patient had a past pathological history of fully treated syphilis (2018) and perianal condyloma with a surgical resection on 09/2017 and 02/2018. In the family history, it was reported that a maternal uncle died of systemic sclerosis. In the social context, the young person denied drinking alcohol and previous or current smoking.
On physical examination, there were no lentiginous skin areas or blue nevi; however, wide violet streaks were observed on the upper limbs and abdomen, with plethora and increased fat in the temporal facial region and hump (Fig. 2a, b), limb ecchymosis, hypotrophy of the arms and thighs, central obesity and kyphoscoliosis. Systemic blood pressure (sitting) was 150 × 90 mmHg, BMI was 26.09 kg/m2, and waist circumference was 99 cm, with no reported reduction in height, maintained at 1.55 m.
Fig. 2 
Changes in the physical examination. a Violet streaks on the upper limbs, b Violet streaks on abdomen
An investigation of secondary causes for osteoporosis was initiated, with the following laboratory test results (Table 2).
Table 2 Laboratory tests Computed tomography of the abdomen with adrenal protocol performed on 08/13/2020 characterized isodense nodular formation in the body of the left adrenal and in the lateral portion of the right adrenal, measuring 1.5 cm and 0.6 cm, respectively. The lesions had attenuation of approximately 30 HU, showing enhancement by intravenous contrast, with an indeterminate washout pattern in the late phase after contrast (< 60%) (Fig. 3).
Fig. 3 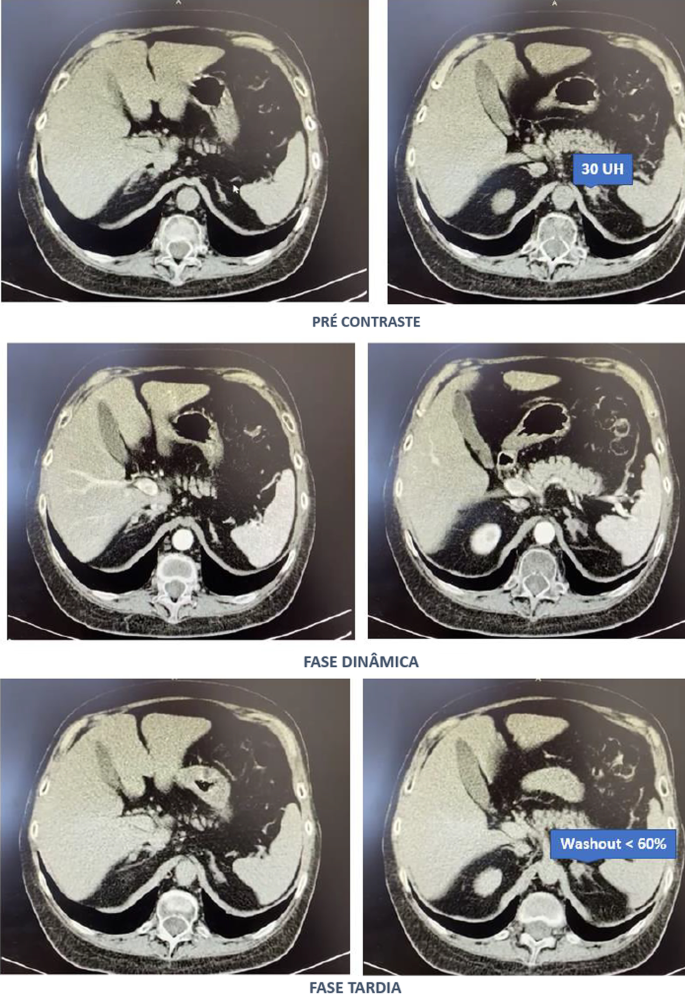
Computed tomography abdomen with adrenal protocol
After contact with the interventional radiology of the Hospital of Federal University of Juiz de Fora, catheterization of adrenal veins was performed on 10/2020; however, it was not possible to perform adequate lesion characterization due to obtaining serum cortisol levels that extrapolated the dilutional upper limit of the method (Table 3).
Table 3 Adrenal catheterization The calculation of the selectivity index was 6.63 (Reference Value (RV) > 3), confirming the good positioning of the catheter within the vessels during the procedure. The calculated lateralization index was 1.1296 (VR < 3), denoting bilateral hormone production. However, as aldosterone was not collected from a peripheral vein, it was not possible to obtain the contralateral rate and define whether there was contralateral suppression of aldosterone production [5].
Due to pending diagnoses for a better therapeutic decision and Cushing’s syndrome in clear evolution and causing organic damage, it was decided, after catheterization, to make changes in the patient’s drug prescription. Ketoconazole 400 mg per day was started, the dose of vitamin D was increased to 14,000 IU per week, and ramipril 5 mg per day was prescribed due to secondary hypertension. In addition, given the severity of osteoporosis, it was decided to replace previously prescribed alendronate with zoledronic acid.
Magnetic resonance imaging of the upper abdomen was performed on 06/19/2021, which demonstrated lobulated nodular thickening in the left adrenal gland with areas of decreased signal intensity in the T1 out-phase sequence, denoting the presence of fat, and homogeneous enhancement using contrast, measuring approximately 1.7 × 1.5 × 1.3 cm, suggestive of an adenoma. There was also a small nodular thickening in the lateral arm of the right adrenal, measuring approximately 0.8 × 0.6 cm, which was difficult to characterize due to its small dimensions and nonspecific appearance.
PPNAD or carcinoma became an important etiological hypothesis for the case described when comparing the epidemiology in a young man and the clinical-laboratory-imaging findings of the differential diagnoses. According to a dialog with the patient and family, the group of experts opted for unilateral glandular surgical resection on the left side (11/11/2021), where more significant changes were visualized, as there was a possibility of malignancy in a young patient and to avoid a definitive adrenal insufficiency condition because of bilateral adrenalectomy. This would first allow the analysis of the material and follow-up of the evolution of the condition with the permanence of the contralateral gland.
In the macroscopic analysis of the adrenalectomy specimen, adrenal tissue weighing 20 g and measuring 9.3 × 5.5 × 2.0 cm was described, completely surrounded by adipose tissue. The gland has a multinodular surface and varies between 0.2 and 1.6 cm in thickness, showing a cortex of 0.1 cm in thickness and a medulla of 1.5 cm in thickness (Fig. 4).
Fig. 4 
Left adrenal
The microscopic analysis described the expansion of the zona fasciculate, with the formation of multiple nonencapsulated nodules composed of polygonal cells with ample and eosinophilic cytoplasm and frequent depletion of intracytoplasmic lipid content. No areas of necrosis or mitotic activity were observed. The histopathological picture is suggestive of cortical pigmented micronodular hyperplasia of the adrenal gland.
For the final etiological definition and an indication of contralateral adrenalectomy, which could be unnecessary and would avoid chronic corticosteroid therapy, or else, it would be necessary to protect the patient from future complications with the maintenance of the disease in the right adrenal gland, it would be essential to search for mutations in the PRKAR1A, PDE11A, PDE8B and PRKACA genes [15]; however, such genetic analysis is not yet widely available, and the impossibility of carrying it out at the local level did not allow a complete conclusion of the case.
Discussion
Through the clinical picture presented and the research of several secondary causes for osteoporosis, it was possible to arrive at the diagnosis of Cushing syndrome [6]. There was symptomatic independent ACTH hypercortisolism, manifested by typical phenotypic changes, severe secondary osteoporosis, and arterial hypertension in a young patient.
The diagnosis of Cushing’s syndrome is always challenging, given the presence of confounding factors such as the following:
-
Physiological states of hypercortisolism—pseudo Cushing (strenuous exercise, pregnancy, uncontrolled diabetes, sleep apnea, chronic pain, alcohol withdrawal, psychiatric disorders, stress, obesity, glucocorticoid resistance syndromes);
-
Cyclic or mild—subclinical Cushing’s pictures;
-
Frequent and, even unknown, short- and long-term use of corticosteroids under different presentations;
-
Increase in the general population incidence of diabetes and obesity;
-
Screening tests with singularities for collection and individualized for different patient profiles.
It is important to note that the basal morning cortisol measurement is not the ideal test to assess hypercortisolism and is better applied to the assessment of adrenal insufficiency. However, the hypercortisolism of the case was unequivocal, and this test was also shown to be altered several times. As no test is 100% accurate, the current guidelines suggest the use of at least two first-line functional tests that focus on different aspects of the pathophysiology of the hypothalamic‒pituitary‒adrenal axis to confirm the hypercortisolism state: 24-hours cortisol, nocturnal salivary cortisol, morning serum cortisol after suppression with 1 mg of dexamethasone or after Liddle 1. Given that night-time salivary cortisol would require hospitalization, the other suggested tests were chosen, which are easier to perform in this context [7, 8].
Subsequently, tests were performed to determine the cause of hypercortisolism, such as serum ACTH levels and adrenal CT. The suppressed ACTH denoted the independence of its action. CT showed bilateral adrenal nodules with more severe features: solid lesion, attenuation > 10 UI on noncontrast images, and contrast washout speed < 60% in 10 minutes. In this case, it is essential to make a broad clinical decision and dialog with the patient to weigh and understand the risks and benefits of surgical treatment [9].
Among the main diagnostic hypotheses for the differential diagnosis of bilateral adrenal hyperplasia are primary bilateral macronodular adrenal hyperplasia, McCune–Albright syndrome (MAS) and bilateral primary pigmented nodular hyperplasia (PPNAD) isolated or associated with Carney’s complex. Another possibility would be bilateral adrenocorticotropic hormone (ACTH)-dependent macronodular hyperplasia secondary to long-term adrenal stimulation in patients with Cushing’s disease (ACTH-secreting pituitary tumor) or ectopic ACTH production, but the present case did not present with ACTH elevation.
Primary macronodular adrenal hyperplasia (nodules > 1 cm) predominates in women aged 50–60 years and may also be detected in early childhood (before 5 years) in the context of McCune–Albright syndrome. Most cases are considered sporadic; however, there are now several reports of familial cases whose presentation suggests autosomal dominant transmission. Several pathogenic molecular causes were identified in the table, indicating that it is a heterogeneous disease [10]. The pathophysiology occurs through the expression of anomalous ectopic hormone receptors or amplified eutopic receptors in the adrenals. It usually manifests in an insidious and subclinical way, with cortisol secretion mediated through receptors for gastric inhibitory peptide (GIP), vasopressin (ADH), catecholamines, interleukin 1 (IL-1), leptin, luteinizing hormone (LH), serotonin or others. Nodular development is not always synchronous or multiple; thus, hypercortisolism only manifests when there is a considerable increase in the number of adrenocortical cells, with severe steroidogenesis observed by cortisoluria greater than 3 times the upper limit of normal. Patients with mild Cushing’s syndrome should undergo screening protocols to identify aberrant receptors, as this may alter the therapeutic strategy. If there is evidence of abnormal receptors, treatment with beta-blockers is suggested for patients with beta-adrenergic receptors or with gonadotropin-releasing hormone (GnRH) agonists (and sex steroid replacement) for patients with LH/hCG receptors. In patients in whom aberrant hormone receptors are not present or for whom no specific pharmacological blockade is available or effective, the definitive treatment is bilateral adrenalectomy, which is known to make the patient dependent on chronic corticosteroid therapy [11]. Studies have shown the effectiveness of unilateral surgery in the medium and long term, opting for the resection of the adrenal gland of greater volume and nodularity by CT, regardless of the values obtained by catheterization of adrenal veins, but with the possibility of persistence or recurrence in the contralateral gland. Another possibility would be total unilateral adrenalectomy associated with subtotal contralateral adrenalectomy [12].
In McCune–Albright syndrome (MAS), there are activating mutations in the G-protein GNAS1 gene, generating autonomic hyperfunction of several tissues, endocrine or not, and there may be, for example, a constant stimulus similar to ACTH on the adrenal gland. In this case, pituitary levels of ACTH are suppressed, and adrenal adenomas with Cushing’s syndrome appear. Hypercortisolism may occur as an isolated manifestation of the syndrome or be associated with the triad composed of polyostotic fibrous dysplasia, café au lait spots with irregular borders and gonadal hyperfunction with peripheral precocious puberty. The natural history of Cushing’s syndrome in McCune-Albright syndrome (MAS) is heterogeneous, with some children evolving with spontaneous resolution of hypercortisolism, while others have a more severe condition, eventually requiring bilateral adrenalectomy [13].
PPNAD predominates in females, in people younger than 30 years, multiple and small (< 6 mm) bilateral pigmented nodules (surrounded by atrophied cortex), which can reach 1.5 cm in adulthood, with family genetic inheritance (66%) or sporadic inheritance (33%), and as part of the Carney complex reported in 40% of cases. In 70% of cases, inactivating mutations are identified in the PKA regulatory 1-alpha subunit (PRKAR1A), a tumor suppressor gene [14]. Osteoporosis is often associated with this condition [15]. One test that can distinguish patients with PPNAD from other primary adrenocortical lesions is cortisoluria after sequential suppression with low- and high-dose dexamethasone. In contrast to most patients with primary adrenocortical disease, who demonstrate no change in urinary cortisol, 70% of PPNAD patients have a paradoxical increase in urinary cortisol excretion [16]. The treatment of choice for PPNAD is bilateral adrenalectomy due to the high recurrence rate for primary adrenal disease [17].
Carney complex is a multiple neoplastic syndrome with autosomal dominant transmission, characterized by freckle-like cutaneous hyperpigmentation (lentiginosis), endocrine tumors [(PPNAD), testicular and/or thyroid tumors and acromegaly] and nonendocrine tumors, including cutaneous, cardiac, mammary, and osteochondral myxomas, among others. In the above case, the transthoracic echocardiogram of the patient on 03/18/2021 showed cavities of normal dimensions, preserved systolic and diastolic functions, no valve changes and no lentiginous skin areas and blue nevi, making the diagnosis of the syndrome less likely. The definitive diagnosis of Carney requires two or more main manifestations. Several related clinical components may suggest the diagnosis but not define it. The diagnosis can also be made if a key criterion is present and a first-degree relative has Carney or an inactivating mutation of the gene encoding PRKAR1A [18].
The adenoma is usually small in size (< 3 cm), similar to the nodules in this case; however, it is usually unilateral, with an insidious and mild evolution, especially in adult women over 35 years of age, producing only 1 steroid class. Carcinomas are usually large (> 6 cm), and only 10% are bilateral. They should be suspected mainly when the tumor presents with hypercortisolism associated with hyperandrogenism. They have a bimodal age distribution, with peaks in childhood and adolescence, as well as at the end of life [3].
Conclusion
Early identification of Cushing’s syndrome, with measures based on the assessment of risks and benefits, remains the best way to prevent progression and reduce morbidity [2]. After 6 months of drug inhibition of steroidogenesis, blood pressure control and anti-osteoporotic therapy, the objective was to minimize the levels and deleterious metabolic effects of hypercortisolism, which could also harm the surgical procedure in the short and long term through infections, dehiscence, nonimmediate bed mobilization and cardiovascular events. Unilateral adrenalectomy was chosen, given the possibility of malignancy in a young patient and to avoid definitive surgical adrenal insufficiency if the adrenalectomy was bilateral. Despite the unavailability of genetic analysis for a precise etiological definition, it is possible to take efficient measures to avoid unnecessary consequences or damage.
Availability of data and materials
All data generated or analysed during this study are included in this published article [and its Additional file 1]. The datasets generated and/or analysed during the current study are available in the link https://ufjfedubr-my.sharepoint.com/:v:/g/personal/barbara_reis_ufjf_edu_br/EVpIR005sPZGlQvMJhIwSaUB0Hig4KOjhkG4D4cMggUwHA?e=Dk8tng.
Abbreviations
- ACTH:
-
Adrenocorticotropic hormone
- PPNAD:
-
Bilateral primary pigmented nodular hyperplasia
- DXA:
-
Dual energy X-ray absorptiometry
- GIP:
-
Gastric inhibitory peptide
- GnRH:
-
Gonadotropin-releasing hormone
- IL-1:
-
Interleukin 1
- BMD:
-
Low bone mineral density
- LH:
-
Luteinizing hormone
- MAS:
-
McCune–Albright syndrome
- PRKAR1A:
-
PKA regulatory 1-alpha subunit
- ADH:
-
Vasopressin
References
-
Pedro AO, Plapler PG, Szejnfeld VL. Manual brasileiro de osteoporose: orientações práticas para os profissionais de saúde. 1st ed. São Paulo: Editora Clannad; 2021. ISBN 978-65-89832-00-3.
-
Naguib R, Elkemary EZ, Elsharkawi KM. The severity of bone loss: a comparison between Cushing’s disease and Cushing’s syndrome. J Endocrinol Metab. 2023;13(1):33–8. https://doi.org/10.14740/jem857.
-
Vilar L, et al. Endocrinologia Clínica. 6th ed. Rio de Janeiro: Guanabara Koogan; 2016.
-
Wang D, Dang CX, Hao YX, Yu X, Liu PF, Li JS. Relationship between osteoporosis and Cushing syndrome based on bioinformatics. Medicine (Baltimore). 2022;101(43): e31283.
-
Williams TA, Reincke M. Management of Endocrine Disease: diagnosis and management of primary aldosteronism: the Endocrine Society guideline 2016 revisited. Eur J Endocrinol. 2018;179(1):R19–29. https://doi.org/10.1530/EJE-17-0990.
-
Compston J, Cooper A, Cooper C, Gittoes N, Gregson C, Harvey N, National Osteoporosis Guideline Group (NOGG), et al. UK clinical guideline for the prevention and treatment of osteoporosis. Arch Osteoporos. 2017;12(1):43. https://doi.org/10.1007/s11657-017-0324-5.
-
Nieman LK. Diagnosis of Cushing’s syndrome in the modern era. Endocrinol Metab Clin N Am. 2018;47(2):259–73. https://doi.org/10.1016/j.ecl.2018.02.001.
-
Herr K, Muglia VF, Koff WJ, Westphalen AC. Imaging of the adrenal gland lesions. Radiol Bras. 2014;47(4):228–39. https://doi.org/10.1590/0100-3984.2013.1762.
-
Hsiao HP, Kirschner LS, Bourdeau I, Keil MF, Boikos SA, Verma S, et al. Clinical and genetic heterogeneity, overlap with other tumor syndromes, and atypical glucocorticoid hormone secretion in adrenocorticotropin-independent macronodular adrenal hyperplasia compared with other adrenocortical tumors. J Clin Endocrinol Metab. 2009;94(8):2930–7. https://doi.org/10.1210/jc.2009-0516.
-
Mircescu H, Jilwan J, N’Diaye N, et al. Are ectopic or abnormal membrane hormone receptors frequently present in adrenal Cushing’s syndrome? J Clin Endocrinol Metab. 2000;85(10):3531–6. https://doi.org/10.1210/jcem.85.10.6865.
-
Miller BS, Auchus RJ. Evaluation and treatment of patients with hypercortisolism: a review. JAMA Surg. 2020;155(12):1152–9. https://doi.org/10.1001/jamasurg.2020.3280.
-
Haddad NG, Eugster EA. Peripheral precocious puberty including congenital adrenal hyperplasia: causes, consequences, management and outcomes. Best Pract Res Clin Endocrinol Metab. 2019;33(3):101273. https://doi.org/10.1016/j.beem.2019.04.007.
-
Bonnet-Serrano F, Bertherat J. Genetics of tumors of the adrenal cortex. Endocr Relat Cancer. 2018;25(3):R131–52. https://doi.org/10.1530/ERC-17-0361.
-
Carney JA, Young WF Jr. Primary pigmented nodular adrenocortical disease and its associated conditions. Endocrinologist. 1992;2:6.
-
Stratakis CA, Sarlis N, Kirschner LS, Carney JA, Doppman JL, Nieman LK, et al. Paradoxical response to dexamethasone in the diagnosis of primary pigmented nodular adrenocortical disease. Ann Intern Med. 1999;131(8):585–91. https://doi.org/10.7326/0003-4819-131-8-199910190-00006.
-
Powell AC, Stratakis CA, Patronas NJ, Steinberg SM, Batista D, Alexander HR, et al. Operative management of Cushing syndrome secondary to micronodular adrenal hyperplasia. Surgery. 2008;143(6):750–8. https://doi.org/10.1016/j.surg.2008.03.022.
-
Almeida MQ, Stratakis CA. Carney complex and other conditions associated with micronodular adrenal hyperplasias. Best Pract Res Clin Endocrinol Metab. 2010;24(6):907–14. https://doi.org/10.1016/j.beem.2010.10.006.
-
Hannah-Shmouni F, Stratakis CA. A gene-based classification of primary adrenocortical hyperplasias. Horm Metab Res. 2020;52(3):133–41. https://doi.org/10.1055/a-1107-2972.
Acknowledgements
Not applicable.
Funding
Not applicable.
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Surgical removal of adrenal gland.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
From https://jmedicalcasereports.biomedcentral.com/articles/10.1186/s13256-023-03968-0
-
1
-
-
Abstract
The association between empty sella turcica (EST) syndrome and Cushing's disease has been rarely reported. It is plausible to hypothesize that EST syndrome in association with Cushing's disease can be attributed to intracranial hypertension. In this case report, we present a 47-year-old male patient who presented with weight loss, fatigue, easy bruising, acanthosis nigricans, and skin creases hyperpigmentation. Investigations revealed hypokalemia and confirmed the diagnosis of Cushing's disease. Magnetic resonance imaging (MRI) brain showed a partial EST syndrome and a new pituitary nodule as compared with previous brain imaging. Transsphenoidal surgery was pursued and was complicated by cerebrospinal fluid leakage. This case reflects the rare association of EST syndrome and Cushing's disease, suggesting the increased risk of postoperative complications in this setting and the diagnostic challenge that EST syndrome imposes. We review the literature for a possible mechanism of this association.
Introduction
Cushing's disease is commonly caused by an adrenocorticotropic hormone (ACTH)-producing pituitary adenoma, which can be very challenging to be seen on brain magnetic resonance imaging (MRI) [1]. Empty sella turcica (EST) syndrome is a radiological diagnosis of apparently empty turcica secondary to outpouching of the arachnoid mater into the turcica, which can be attributed to intracranial hypertension (ICHTN). This can make the visual diagnosis of pituitary adenoma even more challenging in clinical practice. ICHTN has been also associated with Cushing's disease and might explain this infrequent association between EST and Cushing's disease [1]. EST syndrome can be either partial or complete, primary or secondary and has been seen infrequently with Cushing's disease. In this setting, not only that it is likely to obscure an underlying pituitary lesion, but also it does contribute to the risk of postoperative complications [2].
Case Presentation
A 47-year-old male presented to the emergency department (ED) with slowly progressive generalized limb muscle weakness affecting both distal and proximal muscles over a few weeks and gait instability for three days prior to presentation. He also reported unintentional 40 pounds weight loss over the previous four months. Past medical history was significant for type II diabetes mellitus, hypothyroidism, hypertension, and dyslipidemia. In the ED, vital signs included a blood pressure of 140/90 mmHg, a heart rate of 66 beats per minute, a respiratory rate of 16 cycles per minute, and SpO2 of 97% on room air. Body mass index has decreased to 22 kg/m2 from a baseline of 26 kg/m2 one month prior. On the physical exam, he exhibited cachexia, easy bruising, acanthosis nigricans, and hyperpigmentation of skin creases. All other systems were negative. Complete metabolic panel and complete blood count were obtained showing hyperglycemia of 311 mg/dl, see Table 1. Further lab evaluation showed elevated salivary cortisol at 2.96 microgram/dl (reference range 0.094-1.551 mcg/dl), elevated 24-hour urinary free cortisol at 156 mcg/24 hour (reference 10-100 mcg/24h), positive overnight dexamethasone suppression test with serum cortisol at 2.8 mcg/dl (reference more than 2 mcg/dl), negative anti-adrenal antibodies, normal aldosterone, and elevated dehydroepiandrostenedione at 401 mcg/dl (reference 32-240 mcg/dl), with lack of suppression of the ACTH level at 35.1 pg/ml (reference 10-60 pg/ml). This confirmed the diagnosis of Cushing's disease.
Variable Finding Reference Random glucose 311 Less than 200 mg/dl Sodium 141 137-145 mmol/L Potassium 2.5 3.5-5.1 mmol/L Chloride 96 98-107 mmol/L Bicarbonate 32 22-30 mmol/L Blood urea nitrogen 32 9-20 mg/dl Creatinine 0.52 0.66-1.25 mg/dl Calcium 8.7 8.6-10.3 mg/dl Total protein 5.5 6.5-8.5 g/dl Albumin 3.3 3.5-5 g/dl Total bilirubin 0.6 0.2-1.3 mg/dl Alkaline phosphatase 115 38-126 U/L Aspartate transaminase 17 17-59 U/L Alanine transaminase 39 Less than 49 U/L White blood cell count 10x10^3 cells/mcl 4-10x1063 cells/mcl Hemoglobin 15.3 13.7-17.5 g/dl Platelet 281 150-400x10^3 cells/mcl Table 1: Lab Findings
Computed tomography (CT) scan of the head was unremarkable. CT scan of the chest was also unremarkable. CT scan of abdomen and pelvis showed no adrenal mass. Ultrasound of the kidneys was unremarkable. Pituitary MRI brain protocol for adenoma showed a partial EST, shortening within neurohypophysis and a new 10 x 8 x 4 mm nodule along the floor of pituitary sella as compared to MRI four years ago (Figure 1).
-Brain")
Figure 1: Magnetic Resonance Imaging (MRI) Brain
MRI brain showing partially empty sella turcica syndrome ( black arrow) with a small nodule at the floor of the turcica (white arrow).
The diagnosis of Cushing’s disease was confirmed, and the patient underwent trans-sphenoidal resection of pituitary adenoma. Histological examination showed positive CAM 5.2, positive chromogranin, and ACTH immunostains. The patient presented to the ED five days after discharge home. He stated that he noticed drainage from the nose that transitioned from bloody to clear fluid and has been increasing in quantity for two days with associated intermittent headaches since the surgery. He was afebrile with stable vital signs. No signs of infection were noted on basic labs. These were significant only for mild asymptomatic hyponatremia of 131 mmol/L and hypokalemia of 3.3 mmol/L. The patient was diagnosed with cerebrospinal fluid (CSF) leakage and had a lumbar drain trial. The trial was unsuccessful after several days, and the patient underwent a transnasal endoscopic repair of CSF rhinorrhea using nasoseptal flaps. At an outpatient follow-up one month and three months after the surgery, prior lab abnormalities including hypokalemia, hyponatremia, and hyperglycemia resolved. No further evidence of CSF leakage was appreciated, and he remained asymptomatic.
Discussion
EST syndrome is characterized by herniation of the subarachnoid space into the intrasellar space with compression of the pituitary gland into the posteroinferior wall [3]. This is likely to obscure the presence of underlying pituitary mass. The incidence of EST syndrome in the general population is estimated at 20%. The association between EST syndrome and Cushing's disease has been reported infrequently. A retrospective study of 68 patients with Cushing's disease found that 16% of these have EST syndrome [3].
Cushing's disease usually results from pituitary adenomas secreting ACTH, and even the smallest microadenomas can produce a systemic disease. These microadenomas can be very difficult to recognize on brain MRI [4]. This is complicated in EST syndrome and even further with the possibility of ectopic ACTH production. A retrospective study of 197 patients diagnosed with Cushing's disease concluded that EST syndrome is associated with higher prevalence of MRI-negative Cushing's disease. This was attributed to ICHTN and pituitary gland compression [1]. Although surgery is curative in 70-90% of cases, EST syndrome was found to have higher risk of postoperative complications among those with Cushing's disease including diabetes insipidus, hypopituitarism, and CSF leakage [3]. This is usually because in the case of MRI-negative Cushing's disease with total EST syndrome, empiric surgical exploration is sought after inferior petrosal sampling confirms the pituitary origin of excess ACTH, and postoperative remission indicates adequate tumor resection [2]. This entails a higher chance of uncertainty and injury to healthy pituitary tissue.
EST syndrome can be either primarily due to defects in the sellar diaphragm or anatomical variant or secondary to ICHTN. EST syndrome has been reported in association with many conditions associated with elevated intracranial pressure including tumors, thrombosis, meningitis, hydrocephalus, and Arnold-Chiari malformation [5]. Reversal of EST syndrome has been reported in those with idiopathic ICHTN with therapy by acetazolamide, ventriculoperitoneal shunt, and lumbar puncture [6,7]. A study has shown correlation between CSF circulation impairment or blockage and EST syndrome [8]. The incidence of EST syndrome in association with symptomatic intracranial hypertension is variable and ranges from 2.5% for total EST syndrome to 94% for partial EST syndrome [9]. Impaired CSF circulation and dynamics have been reported in 77% of patients with EST syndrome [10]. In addition to intracranial hypertension, EST syndrome has also been described in association with obesity, meningioma, pediatric nevoid basal cell carcinoma, therapy for growth hormone deficiency and even in healthy individuals [9]. Lack of symptoms of intracranial hypertension in this patient does not rule it out as intracranial hypertension in EST syndrome represents a spectrum that ranges from asymptomatic, milder intracranial hypertension to symptomatic intracranial hypertension with headache, visual disturbance, and papilledema [10]. This explains the fact that only 8-14% of EST syndrome progress to symptomatic ICHTN, while symptomatic ICHTN has been associated with EST syndrome in 94% of cases.
ICHTN has been seen in association with disturbance of the hypothalamic-pituitary-adrenal axis. This has been reported after surgical and medical treatment of Cushing's disease, withdrawal of long-term steroid therapy, initial presentation of Addison’s disease, or relative glucocorticoids deficiency [11]. Cortisol excess increases CSF production and reduces its absorption, hence increasing intracranial pressure [12]. Another possible mechanism is the expression of both mineralocorticoid responsive epithelial sodium channel receptors on the basolateral membrane of the CSF producing epithelial cells of the choroid plexus as well as the expression of 11-beta hydroxysteroid dehydrogenase type 1 enzyme, which is a bidirectional enzyme that mainly functions to convert the inactive cortisone to active cortisol. These mechanisms play a role in maintaining the balance between CSF production and absorption [13,14].
In this case, the patient presented some clinical findings that are rarely associated with Cushing's disease, combined with a radiological feature that masked the true diagnosis. Our patient presented with significant weight loss, rather than central obesity, which is normally associated with Cushing’s disease. Although possible, the increase in ACTH due to Cushing's disease is not sufficient to cause hyperpigmentation, which is a classical finding of Addison's disease, where the entire adrenal cortex is usually affected due to an autoimmune destruction; however, the zona glomerulosa of the adrenal cortex produces aldosterone and its deficiency would lead to hyperkalemia [15]. Our patient presented with both hyperpigmentation and hypokalemia.
Conclusions
EST syndrome is an uncommon radiological finding of apparently EST that has been reported in association with ICHTN. The latter has also been seen in association with Cushing's disease/syndrome. This is likely to result from glucocorticoid excess-induced change in CSF flow dynamics. EST has been infrequently described in association with Cushing's disease. This association has a clinical implication as it is likely to obscure the visualization of pituitary lesions responsible for Cushing's disease, contribute to diagnostic uncertainty, and increase the risk of healthy pituitary tissue injury and the risk of postoperative complications including CSF leakage.
References
- Himes BT, Bhargav AG, Brown DA, Kaufmann TJ, Bancos I, Van Gompel JJ: Does pituitary compression/empty sella syndrome contribute to MRI-negative Cushing's disease? A single-institution experience. Neurosurg Focus. 2020, 48:E3. 10.3171/2020.3.FOCUS2084
- Sun Y, Sun Q, Fan C, et al.: Diagnosis and therapy for Cushing's disease with negative dynamic MRI finding: a single-centre experience. Clin Endocrinol (Oxf). 2012, 76:868-76. 10.1111/j.1365-2265.2011.04279.x
- Manavela MP, Goodall CM, Katz SB, Moncet D, Bruno OD: The association of Cushing's disease and primary empty sella turcica. Pituitary. 2001, 4:145-51. 10.1023/a:1015310806063
- Chatain GP, Patronas N, Smirniotopoulos JG, et al.: Potential utility of FLAIR in MRI-negative Cushing's disease. J Neurosurg. 2018, 129:620-8. 10.3171/2017.4.JNS17234
- Friedman DI, Jacobson DM: Diagnostic criteria for idiopathic intracranial hypertension. Neurology. 2002, 59:1492-5. 10.1212/01.wnl.0000029570.69134.1b
- Triggiani V, Giagulli VA, Moschetta M, Guastamacchia E: An unusual case of reversible empty sella. Endocr Metab Immune Disord Drug Targets. 2016, 16:154-6. 10.2174/1871530315666151001141507
- Wind JJ, Lonser RR, Nieman LK, DeVroom HL, Chang R, Oldfield EH: The lateralization accuracy of inferior petrosal sinus sampling in 501 patients with Cushing's disease. J Clin Endocrinol Metab. 2013, 98:2285-93. 10.1210/jc.2012-3943
- Brismar K, Bergstrand G: CSF circulation in subjects with the empty sella syndrome. Neuroradiology. 1981, 21:167-75. 10.1007/BF00367338
- Ranganathan S, Lee SH, Checkver A, Sklar E, Lam BL, Danton GH, Alperin N: Magnetic resonance imaging finding of empty sella in obesity related idiopathic intracranial hypertension is associated with enlarged sella turcica. Neuroradiology. 2013, 55:955-61. 10.1007/s00234-013-1207-0
- Maira G, Anile C, Mangiola A: Primary empty sella syndrome in a series of 142 patients. J Neurosurg. 2005, 103:831-6. 10.3171/jns.2005.103.5.0831
- Zada G, Tirosh A, Kaiser UB, Laws ER, Woodmansee WW: Cushing's disease and idiopathic intracranial hypertension: case report and review of underlying pathophysiological mechanisms. J Clin Endocrinol Metab. 2010, 95:4850-4. 10.1210/jc.2010-0896
- Sinclair AJ, Ball AK, Burdon MA, Clarke CE, Stewart PM, Curnow SJ, Rauz S: Exploring the pathogenesis of IIH: an inflammatory perspective. J Neuroimmunol. 2008, 201:212-20. 10.1016/j.jneuroim.2008.06.029
- Sinclair AJ, Onyimba CU, Khosla P, et al.: Corticosteroids, 11beta-hydroxysteroid dehydrogenase isozymes and the rabbit choroid plexus. J Neuroendocrinol. 2007, 19:614-20. 10.1111/j.1365-2826.2007.01569.x
- Amin MS, Wang HW, Reza E, Whitman SC, Tuana BS, Leenen FH: Distribution of epithelial sodium channels and mineralocorticoid receptors in cardiovascular regulatory centers in rat brain. Am J Physiol Regul Integr Comp Physiol. 2005, 289:R1787-97. 10.1152/ajpregu.00063.2005
- Stratakis CA: Skin manifestations of Cushing's syndrome. Rev Endocr Metab Disord. 2016, 17:283-6. 10.1007/s11154-016-9399-3
-
1
-
The Cushing’s Hub Editorial Board announces the final call for the 2023 Cushing’s Hub Competition!
Now is your last chance to submit your favourite clinical scenario for a chance to see it appear as an Interactive Clinical Case on Cushing’s Hub. While any Cushing’s-focused scenario is welcome, submissions that examine interesting presentations of mild-to-severe hypercortisolaemia, or long-term disease outcomes (including health-related quality of life) are particularly encouraged.
The successful entrant will see their clinical scenario developed into a pedagogically enhanced, interactive e-learning module. The winner will also be invited to take part in a short film sequence to introduce their work, which will be promoted at launch worldwide via social media.
Don’t miss a chance to share your work with the endocrine community, click here for details on how to enter.
Closing date for entries 30 June 2023-
1
-
-
I'm glad I didn't know this before my recent knee surgery!
Abstract
Background
Cushing’s syndrome (CS) is a disorder characterized by exposure to supraphysiologic levels of glucocorticoids. The purpose of this study was to evaluate the association between CS and postoperative complication rates following total joint arthroplasty (TJA).
Methods
Patients diagnosed with CS undergoing TJA for degenerative etiologies were identified from a large national database and matched 1:5 to a control cohort using propensity scoring. Propensity score matching resulted in 1,059 total hip arthroplasty (THA) patients with CS matched to 5,295 control THA patients and 1,561 total knee arthroplasty (TKA) patients with CS matched to 7,805 control TKA patients. Rates of medical complications occurring within 90 days of TJA and surgical-related complications occurring within 1 year of TJA were compared using odds ratios.
Results
The THA patients with CS had higher incidences of pulmonary embolism (Odds Ratio (OR) 2.21, P=0.0026), urinary tract infection (OR 1.29, P=0.0417), pneumonia (OR 1.58, P=0.0071), sepsis (OR 1.89, P=0.0134), periprosthetic joint infection (OR 1.45, P=0.0109), and all-cause revision surgery (OR 1.54, P=0.0036). The TKA patients with CS had significantly higher incidences of urinary tract infection (OR 1.34, P=0.0044), pneumonia (OR 1.62, P=0.0042), and dislocation (OR 2.43, P=0.0049) and a lower incidence of manipulation under anesthesia (MUA) (OR 0.63, P=0.0027).
Conclusion
Cushing’s syndrome is associated with early medical- and surgical-related complications following TJA and a reduced incidence of MUA following TKA.
Introduction
Cushing’s syndrome (CS) is characterized by exposure to supraphysiologic levels of glucocorticoids, whether endogenous or exogenous. Chronic exposure to hypercortisolism can lead to the development of comorbidities known to be risk factors for complications following total joint arthroplasty (TJA) including obesity, hypertension, diabetes, hyperlipidemia, and cerebrovascular disease.[1,2] Hypercortisolism is also a known risk factor for the development of osteonecrosis, and there have been several case reports of this disease being caused by endogenous production of corticosteroids.[3, 4, 5, 6, 7, 8] It can therefore be expected that the incidence of arthroplasty procedures among CS patients is likely higher than the general population. It is important to identify and understand patient specific risk factors for complications following TJA. There has been a major push recently to investigate the association between uncommon disorders and complication rates following TJA in order to risk stratify, counsel, and optimize these patients appropriately.[9, 10, 11, 12, 13, 14, 15]
The typical clinical features of CS include increased central adiposity, purple striae, thin skin, fatigue, and proximal atrophy of the upper and lower limbs.[16,17] The most common etiology of endogenous CS is overproduction of adrenocorticotropic hormone (ACTH) from a pituitary adenoma, although ACTH-independent forms of CS may be caused by overproduction of glucocorticoids from the adrenal glands.[2] First-line laboratory tests for the diagnosis of CS include 24-hour urinary free cortisol, late night salivary cortisol, and the dexamethasone suppression test to determine if the negative feedback of the hypothalamic-pituitary-adrenal axis is functioning appropriately.[16] Hypercortisolism associated with CS is known to have a deleterious effect on bone health by decreasing osteoblast function and increasing bone resorption and has been associated with decreased bone mineral density at various sites in the femur including Ward’s triangle, the femoral neck, and the greater trochanter.[18] The effect of these changes in physiology on outcomes following TJA remains unclear. There are few prior case reports describing arthroplasty procedures for CS patients,[3, 4, 5] with one case report of total hip arthroplasty (THA) for femoral head osteonecrosis complicated by pulmonary thromboembolism requiring a 10-day admission to the ICU.[3] However, no large scale studies to date have investigated complication rates following TJA within this patient population. It is therefore important to better understand the risks associated with this pathology. The purpose of this study was to evaluate the association between CS and postoperative complication rates following TJA. We hypothesized that patients who have CS would have increased incidences of early medical- and surgical-related complications.
Section snippets
Methods
This is a retrospective cohort study utilizing the commercially available M151Ortho database via PearlDiver (PearlDiver Inc., Colorado Springs, Colorado). This database contains deidentified records for 151 million patients in the United States in accordance with the Health Insurance Portability and Accountability Act (HIPAA). Patient records were queried using International Classification of Diseases (ICD) codes and Current Procedural Terminology (CPT) codes. This study was deemed exempt from
Results
The THA patients who had CS had significantly higher 90-day incidences of PE (OR 2.21, P=0.0026), UTI (OR 1.29, P=0.0417), pneumonia (OR 1.58, P=0.0071), and sepsis (OR 1.89, P=0.0134) (Table 2). The TKA patients who had CS had significantly higher 90-day incidences of UTI (OR 1.34, P=0.0044) and pneumonia (OR 1.62, P=0.0042) (Table 3). Regarding surgical-related complications, CS patients undergoing THA had significantly higher incidences of PJI (OR 1.45, P=0.0109) and all-cause revision
Discussion
This study revealed that patients who have CS are at increased risk of developing early postoperative complications following TJA. Understanding this risk profile is important for accurate shared decision making between CS patients and their clinicians. Interestingly, CS seems to influence rates of instability and stiffness following TKA as patients in the test cohort were more likely to sustain a dislocation and less likely to undergo MUA. Rates of infectious medical complications including
Conclusion
Cushing’s syndrome is associated with an increased risk of early infectious complications following TJA including UTI, pneumonia, sepsis, and hip PJI and an increased incidence of dislocation following TKA. Interestingly, CS appears to be protective against arthrofibrosis as patients who have CS had lower incidences of MUA following TKA. Clinicians may be guided by this study to accurately risk stratify and counsel patients with CS prior to undergoing TJA.
References (29)
-
A. Lacroix et al.
Cushing’s syndrome
Lancet
(2015) -
H.G. Moore
Total Joint Arthroplasty in Patients With Achondroplasia: Comparison of 90-Day Adverse Events and 5-Year Implant Survival
Arthroplast Today
(2021) -
M.J. Gouzoulis et al.
Hidradenitis Suppurativa Leads to Increased Risk of Wound-Related Complications following Total Joint Arthroplasty
Arthroplast Today
(2022) -
H.G. Moore
Total Hip Arthroplasty in Patients With Cerebral Palsy: A Matched Comparison of 90-Day Adverse Events and 5-Year Implant Survival
J. Arthroplasty
(2021) -
M.W. Cole et al.
The Impact of Celiac Disease on Complication Rates After Total Joint Arthroplasty: A Matched Cohort Study
Arthroplast Today
(2022) -
M.R. Mercier et al.
Outcomes Following Total Hip Arthroplasty in Patients With Postpolio Syndrome: A Matched Cohort Analysis
J. Arthroplasty
(2022) -
M. Barbot et al.
Cushing’s syndrome: Overview of clinical presentation, diagnostic tools and complications
Best Pract. Res. Clin. Endocrinol. Metab
(2020) -
E. Salt
Moderating effects of immunosuppressive medications and risk factors for post-operative joint infection following total joint arthroplasty in patients with rheumatoid arthritis or osteoarthritis
Semin. Arthritis Rheum
(2017) -
R. Gandhi
Predictive risk factors for stiff knees in total knee arthroplasty
J. Arthroplasty
(2006) -
P.E. Scranton
Management of knee pain and stiffness after total knee arthroplasty
J. Arthroplasty
(2001)
-
1
-
A. Lacroix et al.
-
Abstract
Summary
Cushing’s syndrome due to ectopic adrenocorticotropic hormone (ACTH) secretion (EAS) by a pheochromocytoma is a challenging condition. A woman with hypertension and an anamnestic report of a ‘non-secreting’ left adrenal mass developed uncontrolled blood pressure (BP), hyperglycaemia and severe hypokalaemia. ACTH-dependent severe hypercortisolism was ascertained in the absence of Cushingoid features, and a psycho-organic syndrome developed. Brain imaging revealed a splenial lesion of the corpus callosum and a pituitary microadenoma. The adrenal mass displayed high uptake on both 18F-FDG PET/CT and 68Ga-DOTATOC PET/CT; urinary metanephrine levels were greatly increased. The combination of antihypertensive drugs, high-dose potassium infusion, insulin and steroidogenesis inhibitor normalized BP, metabolic parameters and cortisol levels; laparoscopic left adrenalectomy under intravenous hydrocortisone infusion was performed. On combined histology and immunohistochemistry, an ACTH-secreting pheochromocytoma was diagnosed. The patient's clinical condition improved and remission of both hypercortisolism and catecholamine hypersecretion ensued. Brain magnetic resonance imaging showed a reduction of the splenial lesion. Off-therapy BP and metabolic parameters remained normal. The patient was discharged on cortisone replacement therapy for post-surgical hypocortisolism. EAS due to pheochromocytoma displays multifaceted clinical features and requires prompt diagnosis and multidisciplinary management in order to overcome the related severe clinical derangements.
Learning points
-
A small but significant number of cases of adrenocorticotropic hormone (ACTH)-dependent Cushing’s syndrome are caused by ectopic ACTH secretion by neuroendocrine tumours, which is usually associated with severe hypercortisolism causing severe clinical and metabolic derangements.
-
Ectopic ACTH secretion by a pheochromocytoma is exceedingly rare but can be life-threatening, owing to the simultaneous excess of both cortisol and catecholamines.
-
The combination of biochemical and hormonal testing and imaging procedures is mandatory for the diagnosis of ectopic ACTH secretion, and in the presence of an adrenal mass, the possibility of an ACTH-secreting pheochromocytoma should be taken into account.
-
Immediate-acting steroidogenesis inhibitors are required for the treatment of hypercortisolism, and catecholamine excess should also be appropriately managed before surgical removal of the tumour.
-
A multidisciplinary approach is required for the treatment of this challenging entity.
Keywords: Adult; Female; White; Italy; Adrenal; Pituitary; Unique/unexpected symptoms or presentations of a disease; May; 2023Background
Cushing’s syndrome (CS) is a rare endocrine disease characterized by high levels of glucocorticoids; it increases morbidity and mortality due to cardiovascular and infectious diseases (1, 2, 3).
To diagnose CS, adrenocorticotropic hormone (ACTH)-dependent disease must be distinguished from ACTH-independent disease, and pituitary ACTH production from ectopic production. About 20% of ACTH-dependent cases arise from ectopic ACTH secretion (EAS) (2, 3, 4). EAS is most often due to aberrant ACTH production by small-cell lung carcinoma or neuroendocrine tumours originating in the lungs or gastrointestinal tract; this, in turn, strongly increases cortisol production by the adrenal glands (3, 4, 5).
Since the first-line treatment of EAS is the surgical removal of the ectopic ACTH-secreting tumour, its prompt and accurate localization is crucial.
Rapid cortisol reduction by means of immediate-acting steroidogenesis inhibitors (4) is mandatory in order to treat the related endocrine, metabolic and electrolytic derangements. EAS by a pheochromocytoma is exceedingly rare and can be life-threatening.
We describe the case of a woman with hypertension and a known ‘non-secreting’ left adrenal mass, who manifested uncontrolled blood pressure (BP), hyperglycaemia, hypokalaemia and psycho-organic syndrome associated with damage of the splenium of the corpus callosum. These findings were eventually seen to be related to an ACTH-secreting left pheochromocytoma, which was ascertained by hormonal evaluation and morphological and functional imaging assessment and confirmed by histopathology/immunostaining. Hormonal hypersecretion resolved after adrenalectomy and metabolic derangements normalized.
Case presentation
A 72-year-old woman with hypertension was taken to the emergency department because of increased BP (200/100 mm Hg). High BP (190/100 mmHg) was confirmed, whereas oxygen saturation (98%), heart rate (84 bpm) and lung and abdomen examination were normal. Electrocardiogram and chest x-ray were unremarkable. Captopril 50 mg orally, followed by intramuscular clonidine, normalized BP.
The patient looked thin and reported significant weight loss (10 kg) over the previous 6 months; she was on antihypertensive therapy with bisoprolol 5 mg/day and irbesartan 150 mg/day, and ezetimibe 10 mg/day for dyslipidaemia. The patient’s records included a previous diagnosis in another hospital of normofunctioning multinodular goitre and a 2.5 cm-left solid inhomogeneous adrenal mass with well-defined margins, which was found on CT performed 6 years earlier during the work-up for hypertension. On the basis of hormonal data and absent uptake on 123I metaiodobenzylguanidine scintigraphy, the adrenal lesion had been deemed to be non-functioning and follow-up had been advised. Unfortunately, only initial cortisol (15.7 μg/dL) and 24-h urine-free cortisol (UFC) levels (32.5 μg/24 h) were retrievable; both proved normal.
Investigations
Blood chemistry showed neutrophilic leucocytosis, hyperglycaemia with increased glycated haemoglobin, severe hypokalaemia and metabolic alkalosis (Table 1). Potassium infusion (50 mEq in 500 mL saline/24 h) was rapidly started, together with a subcutaneous rapid-acting insulin analogue and prophylactic enoxaparin. The patient experienced mental confusion, hallucinations and restlessness; non-enhanced computed tomography (CT) of the brain revealed a hypodense area of the splenium of the corpus callosum, possibly due to metabolic damage (Fig. 1A).

Figure 1 Non-enhanced CT showing a hypodense area of the splenium of the corpus callosum (arrows), without mass effect (A, axial view). Contrast-enhanced MR image showing a hypointense pituitary lesion (arrow) which enhances more slowly than normal pituitary parenchyma, deemed suspicious for microadenoma (B, coronal view). FLAIR MR image showing hyperintense signal of the splenium of the corpus callosum (asterisk), which partially extended to the crux of the left fornix (arrow) (C, axial view). As the lesion showed no restricted diffusion on DWI (D, axial view), an ischaemic lesion was excluded. A progressive reduction in the extension of the hyperintense signal in the splenium of the corpus callosum (arrowheads) and in the crux of the left fornix (arrows) was observed on FLAIR MR images (2 months (E); 3 months (F); axial view). CT, computed tomography; DWI, diffusion-weighted imaging; FLAIR, fluid-attenuated inversion recovery; MR, magnetic resonance.
Citation: Endocrinology, Diabetes & Metabolism Case Reports 2023, 2; 10.1530/EDM-22-0308
Table 1Hormonal and biochemical evaluation of patient throughout hospitalization and follow-up.
Normal range On hospital admission After surgery 10 days 2 months 3 months 6 months 9 months 12 months 16 months ACTH (pg/mL) 9–52 551 7 37 50 29.5 26 40.9 52 Morning cortisol† (µg/dL) 7–19.2 63.4 14 5.1 3.5 3.8 4.2 7.2 12.8 After 1 mg overnight dexamethasone ACTH 583 Cortisol 60 DHEAS (µg/dL) 9.4–246 201 24-h urinalysis (µg/24 h) Adrenaline 0–14.9 95.5 Noradrenaline 0–66 1133 Metanephrine 74–297 1927 Normetanephrine 105–354 1133 Chromogranin A 0–108 290 Renin (supine) (µU/mL) 2.4–29 3.9 14.6 Aldosterone (supine) (ng/dL) 3–15 3.4 12.5 LH (mIU/mL)* > 10 0.3 65.8 FSH (mIU/mL)* > 25 1.9 116 PRL (ng/mL) 3–24 13.7 FT4 (ng/dL) 0.9–1.7 1.1 1.2 FT3 (pg/mL) 1.8–4.6 1.1 2.7 TSH (µU/mL) 0.27–4.2 0.23 1.3 PTH (pg/mL) 15–65 166 Calcium (mg/dL) 8.2–10.2 8.2 Calcitonin (pg/mL) 0–10 1 Glycaemia (mg/dL) 60–110 212 69 73 83 Potassium (mEq/L) 3.5–5 2.4 3.3 3.9 4.2 3.7 5 4.4 3.9 Leucocytes (K/µL) 4.0–9.3 15.13 HbA1c (mmol/mol) 20–42 55 30 HCO3− (mEq/L) 22–26 41.8 *For menopausal age; †07:00–10:00 h.
The patient was transferred to the internal medicine ward. Although potassium infusion was increased to 120 mEq/day, serum levels did not normalize; a mineralocorticoid receptor antagonist (potassium canreonate) was therefore introduced, but the effect was partial. In order to control BP, the irbersartan dose was increased (300 mg/day) and amlodipine (10 mg/day) was added.
The combination of severe hypertension, newly occurring diabetes and resistant hypokalaemia prompted us to hypothesize a common endocrine aetiology.
A thorough hormonal array showed very high ACTH and cortisol levels, whereas supine renin and aldosterone levels were in the low-normal range (Table 1). Since our patient proved repeatedly non-compliant with 24-h urine collection, UFC could not be measured.
After an overnight 1 mg dexamethasone suppression test, cortisol levels remained unchanged, whereas ACTH levels slightly increased (Table 1). Notably, the patient showed no Cushingoid features. Gonadotropin levels were inappropriately low for the patient’s age; FT4 levels were normal, whereas FT3 and thyroid-stimulating hormone (TSH) levels were reduced and calcitonin levels were normal (Table 1). HbA1c levels were increased (Table 1).
Finally, secondary hyperparathyroidism, associated with low-normal calcium levels and reduced vitamin D levels, was found (Table 1).
Brain contrast-enhanced magnetic resonance (MR) imaging revealed a 5-mm median posterior pituitary microadenoma (Fig. 1B) and a hyperintense lesion of the splenium of the corpus callosum (Fig. 1C). Diffusion-weighted MR images of the lesion showed no restricted diffusion (Fig. 1D), thus excluding an ischaemic origin. Petrosal venous sampling for ACTH determination at baseline and after CRH stimulation was excluded, as it was deemed a high-risk procedure, given the patient's poor condition.
Since the ACTH and cortisol levels were greatly increased and were associated with severe hypokalaemia, EAS was hypothesized; total-body contrast-enhanced CT revealed the left adrenal mass (3 cm), which showed regular margins and heterogeneous enhancement (Fig. 2A and B) and measured 25 Hounsfield units. There was no evidence of adrenal hyperplasia in the contralateral adrenal gland. The adrenal mass showed intense tracer uptake on both 18F-FDG PET/CT (Fig. 2C and D), suggestive of adrenal malignancy or functioning tumour, and 68Ga-DOTATOC PET/CT (Fig. 3), which is characteristic of a neuroendocrine lesion. No other sites of suspicious tracer uptake were detected.

Figure 2 Contrast-enhanced abdominal computed tomography showing a 3-cm left adrenal mass (arrow) with well-defined margins and inhomogeneus enhancement, deemed compatible with an adenoma (A, coronal view; B, axial view). The adrenal mass showed high uptake (SUV max: 7.3) on 18F-FDG PET/CT (C, coronal view; D, axial view).
Citation: Endocrinology, Diabetes & Metabolism Case Reports 2023, 2; 10.1530/EDM-22-0308

Figure 3 The left adrenal mass displaying very high uptake (SUV max: 40) on 68Ga-DOTATOC PET/CT (arrow, axial view).
Citation: Endocrinology, Diabetes & Metabolism Case Reports 2023, 2; 10.1530/EDM-22-0308
Bisoprolol was withdrawn, and 24-h urinary catecholamine, metanephrine and normetanephrine levels proved significantly increased, as were chromogranin A levels (Table 1). In sum, an ACTH-secreting pheochromocytoma was suspected and the pituitary microadenoma was deemed a likely incidental finding.
The patient’s mental state worsened, fluctuating from sopor to restlessness, which required parenteral neuroleptics and restraint. An electroencephalogram revealed a specific slowdown of cerebral electrical activity. Following rachicentesis, the cerebrospinal fluid showed pleocytosis (lympho-monocytosis), whereas a culture test and polymerase chain reaction for common neurotropic agents were negative. The neurologist hypothesized a psycho-organic syndrome secondary to severe metabolic derangement. Intravenous ampicillin, acyclovir and B vitamins were empirically started. The patient was transferred to the subintensive unit, where a nasogastric tube and central venous catheter were inserted, and enteral nutrition was started.
Treatment
Ketoconazole was started at a dosage of 200 mg twice daily; both cortisol and ACTH levels significantly decreased over a few days (Fig. 4), with a progressive decrease in glucose levels and normalization of potassium levels and BP on therapy. Subsequently, ketoconazole was titrated to 600 mg/day owing to a new increase in cortisol levels, which eventually normalized (Fig. 4). Of note, ACTH levels partially decreased on ketoconazole treatment (Fig. 4).

Figure 4 ACTH and cortisol levels throughout the patient’s hospitalization and follow-up.
Citation: Endocrinology, Diabetes & Metabolism Case Reports 2023, 2; 10.1530/EDM-22-0308
Doxazosin 2 mg/day was added and the patient's systolic BP blood settled at around 100 mm Hg; after a few days, bisoprolol was restarted. Contrast-enhanced MR showed a partial reduction of the hyperintense splenial lesion (Fig. 1E). Despite the severe clinical condition and the high risks of adrenal surgery, the patient’s relatives strongly requested the procedure and laparoscopic left adrenalectomy was planned. Alpha-blocker and fluid infusion were continued, ketoconazole was withdrawn the day before surgery, and a 100 mg IV bolus of hydrocortisone was administered just before the operation, followed by 200 mg/day, at first in continuous infusion, then as a 100 mg bolus every 8 h. After the removal of the left adrenal mass, noradrenaline infusion was required, owing to the occurrence of severe hypotension.
Outcome and follow-up
Pathology revealed a 2.5 cm reddish-brown encapsulated tumour, which was compatible with pheochromocytoma (Fig. 5A and B); ACTH immunostaining was positive in about 30% of tumour cells (Fig. 5C). This confirmed the diagnostic hypothesis of an ACTH-secreting pheochromocytoma. The tumour was stained for Chromogranin A (Fig. 5D). There were no signs of adrenal cortex hyperplasia in the resected gland. Thorough germinal genetic testing, comprising the commonest pheochromocytoma/paraganglioma genes: CDKN1B, KIF1B, MEN1, RET, SDHA, SDHB, SDHC, SDHD, SDHAF2 and TMEM127, was negative.

Figure 5 Histological images of adrenal pheochromocytoma: the tumour is composed of well-defined nests of cells (‘zellballen’) (A; haematoxylin-eosin stain (HE), ×20) with pleomorphic nuclei with prominent nucleoli, basophilic or granular amphophilic cytoplasm (B; HE, ×40). The mitotic index was low: 1 mitosis per 30 high-power fields, and Ki-67 was 1%. On immunohistochemistry, cytoplasmatic ACTH staining was found in about 30% of tumour cells (C; ×20), whereas most tumour cells were stained for chromogranin A (D; ×20).
Citation: Endocrinology, Diabetes & Metabolism Case Reports 2023, 2; 10.1530/EDM-22-0308
One week after surgery ACTH levels had dropped to a low-normal value: 7 pg/mL, and cortisol levels (before morning hydrocortisone bolus administration) were normal: 14 µg/dL (Fig. 4). The patient’s clinical status slowly improved and the nasogastric tube was removed; intravenous hydrocortisone was carefully tapered until withdrawal and high-dose oral cortisone acetate (62.5 mg/day) was started. This dose was initially required since BP remained low (systolic: 90 mm Hg); thereafter, cortisone was reduced to 37.5 mg/day. Plasma cortisol levels before morning cortisone administration were reduced (Fig. 4). A new MR of the brain showed a further partial reduction of the splenial lesion (Fig. 1F). The patient was discharged with normal off-therapy BP and metabolic parameters.
During follow-up, she fully recovered, and BP and metabolic parameters remained normal. Gonadotropin levels became adequate for the patient’s age, and TSH and renin/aldosterone levels normalized (Table 1). Hypoadrenalism, however, persisted for more than 1 year; as the last hormonal evaluation, 16 months after surgery, showed normal baseline cortisol levels, the cortisone dose was tapered (12.5 mg/day) and further hormonal examination was scheduled (Table 1). ACTH and cortisol levels throughout the patient’s hospitalization and follow-up are shown in Fig. 4.
Discussion
The diagnosis of EAS is challenging and requires two steps: confirmation of increased ACTH and cortisol levels and anatomic distinction from pituitary sources of ACTH overproduction. Besides metabolic derangements (hyperglycaemia, hypertension), EAS-related severe hypercortisolism may cause profound hypokalaemia (3, 4, 5).
In our patient, the combination of worsening hypertension, newly occurring diabetes and resistant hypokalaemia raised the suspicion of a common endocrine cause.
ACTH-dependent severe hypercortisolism was ascertained, and subsequent brain MR revealed a pituitary microadenoma.
The diagnosis of CS requires the combination of two abnormal test results: 24-h UFC, midnight salivary cortisol and/or abnormal 1 mg dexamethasone suppression testing (2, 6). ACTH evaluation (low/normal-high) is fundamental to tailoring the imaging technique.
The very high cortisol levels found in our patient were unchanged after overnight dexamethasone testing, whereas UFC could not be assessed owing to the lack of compliance with urine collection. The accuracy of the UFC assays, however, may be impaired by cortisol precursors and metabolites. Salivary cortisol assessment was not performed since the specific assay is not available in our hospital.
The combination of ACTH-dependent severe hypercortisolism and hypokalaemia prompted us to suspect EAS. The differential diagnosis between pituitary and ectopic ACTH-dependent CS involves high-dose (8 mg) dexamethasone suppression testing, which has relatively low diagnostic accuracy (6). Owing to the patient's very high cortisol levels and severe hypokalaemia, this testing was not performed, on account of the risks of administering corticosteroids in a patient already exposed to excessive levels (6). Furthermore, owing to the increase in ACTH levels observed after overnight dexamethasone testing, we postulated the possible occurrence of glucocorticoid-driven positive feedback on ACTH secretion, which has been described in EAS, including cases of pheochromocytoma (7).
Finally, in the case of EAS suspected of being caused by pheochromocytoma, we do not recommend performing high-dose dexamethasone suppression testing, owing to the risk of triggering a catecholaminergic crisis (8).
The dynamic tests commonly used to distinguish patients with EAS from those with Cushing's disease are the CRH stimulation test and the desmopressin stimulation test, either alone or in combination with CRH testing (6). Owing to the rapid worsening of our patient’s condition, dynamic testing was not done; however, the clinical picture and hormonal/biochemical data were suggestive of EAS.
EAS is mainly (45–50%) due to neuroendocrine tumours, mostly of the lung (small-cell lung cancer and bronchial tumours), thymus or gastrointestinal tract; however, up to 20% of ACTH-secreting tumours remain occult (3, 4, 5).
ACTH-secreting pheochromocytomas are responsible for about 5% of cases of EAS (3, 4, 9, 10). Indeed, this rate ranges widely, from 2.5% (11) to 15% (12), according to the different case series. Patients with EAS due to pheochromocytoma present with severe CS, overt diabetes mellitus, hypertension and hypokalaemia (3); symptoms of catecholamine excess may be unapparent (3), making the diagnosis more challenging.
A recent review of 99 patients with ACTH- and/or CRH-secreting pheochromocytomas found that the vast majority displayed a Cushingoid phenotype (10); by contrast, another review of 24 patients reported that typical Cushingoid features were observed in only 30% of patients, whereas weight loss was a prevalent clinical finding (13). We hypothesized that the significant weight loss reported by our patient was largely due to the hypermetabolic state induced by catecholamines, which directly reduce visceral and subcutaneous fat, as recently reported (14).
Our patient showed no classic stigmata of CS, owing to the rapid onset of severe hypercortisolism (10, 13), whereas she had worsening hypertension and newly occurring diabetes mellitus, which were related to both cortisol and catecholamine hypersecretion; hypokalaemia was deemed to be secondary to severe hypercortisolism. Indeed, greatly increased cortisol levels act on the mineralocorticoid receptors of the distal tubule after saturating 11β-hydroxysteroid dehydrogenase type 2, leading to hypokalaemia (4). Consequently, hypokalaemia is much more common (74–95% of patients) in EAS than in classic Cushing’s disease (10%) (3, 4, 10). This apparent mineralocorticoid excess suppresses renin and aldosterone secretion, as was ascertained in our patient.
In this setting, the most effective way to manage hypokalaemia is to treat the hypercortisolism itself by administering immediate-acting steroidogenesis inhibitors, combined with potassium infusion and a mineralocorticoid receptor-antagonist (e.g. spironolactone) at an appropriate dosage (100–300 mg/day) (4).
In ACTH-secreting pheochromocytoma, cortisol hypersecretion potentiates catecholamine-induced hypertension by stimulating the phenol-etholamine-N-methyl–transferase enzyme, which transforms noradrenaline to adrenaline (4). Indeed, in our patient, the significant ketoconazole-induced reduction in cortisol secretion led to satisfactory BP control on antihypertensive drugs. After the biochemical diagnosis of pheochromocytoma, a selective alpha-blocker was added, and after a few days, a beta-blocker was restarted in order to control reflex tachycardia (15).
Our patient had greatly increased ACTH levels (>500 pg/mL) associated with very high cortisol levels (>60 µg/dL), which, together with the finding of hypokalaemia, prompted us to hypothesize EAS. With regard to these findings, ACTH levels are usually higher (>200 pg/mL) in patients with EAS than in those with CS due to a pituitary adenoma; however, considerable overlapping occurs (3, 11, 16). Most patients with ACTH-secreting pheochromocytomas in those series had ACTH levels >300 pg/mL, and a few had normal ACTH levels (9), thus complicating the diagnosis. In addition, patients with EAS usually have higher cortisol levels than those with ACTH-secreting adenomas (3, 11).
In our patient, the left adrenal mass was deemed the culprit of EAS, and owing to very high urinary metanephrine levels, a pheochromocytoma was suspected.
It can be assumed that the adrenal tumour, which was anamnestically reported as ‘non-secreting’, but on which only part of the initial hormonal data were available, was actually a pheochromocytoma at the time of the first diagnosis but displayed a silent clinical and hormonal behaviour. The mass subsequently showed significant uptake on both 18F-FDG PET/CT and 68Ga-DOTATOC PET/CT (4, 5). It is claimed that 68Ga-DOTATOC PET/CT provides a high grade (90%) of sensitivity and specificity in the diagnosis of tumours that cause EAS (4, 5); nevertheless, a recent systematic review reported much lower sensitivity (64%), which increased to 76% in histologically confirmed cases (17).
In patients with EAS, immediate-acting steroidogenesis inhibitors are required in order to achieve prompt control of severe hypercortisolism (4). Ketoconazole is one of the drugs of choice since it inhibits adrenal steroidogenesis at several steps. In our patient, ketoconazole rapidly reduced cortisol levels to normal values, without causing hepatic toxicity (4). Moreover, ketoconazole proved effective at a moderate dosage (600 mg/day), which falls within the mean literature range (18, 19). However, dosages up to 1200–1600 mg/day are sometimes required in severe cases (usually EAS) (18, 19). Speculatively, our results might reflect an enhanced inhibitory action of ketoconazole at the adrenal level, which was able to override the strong ectopic ACTH stimulation.
In addition, the finding that, following cortisol reduction, ACTH levels paradoxically decreased suggests an additive and direct effect of the drug. This effect has been observed in a few patients with EAS (20) and is supported by in vitro studies showing a direct anti-proliferative and pro-apoptotic effect of ketoconazole on ectopic ACTH secretion by tumours (21). Finally, the reduction in ACTH levels during treatment with steroidogenesis inhibitors prompts us to postulate the presence of glucocorticoid-driven positive feedback on ACTH secretion, as already described in neuroendocrine tumours (7, 20, 21). The coexistence of EAS and ACTH-producing pituitary adenoma is very rare but must be taken into account. In our case, we deemed the pituitary mass found on MR to be a non-secreting microadenoma. This hypothesis was strengthened by the finding that, following exeresis of the ACTH-secreting pheochromocytoma, ACTH normalized, hypercortisolism vanished and pituitary function recovered. These findings suggest that: (i) altered pituitary function at the baseline was secondary to the inhibitory effect of hypercortisolism; (ii) the excessive production of cortisol was driven by ACTH overproduction outside the pituitary gland, specifically within the adrenal gland tumour.
In our patient, a few days after surgery, morning cortisol levels before hydrocortisone bolus administration were ‘normal’. Owing to both the half-life of hydrocortisone (8–12 h) and the supraphysiological dosage used, it is likely that a residual part of the drug, which cross-reacts in the cortisol assay, was still circulating at the time of blood collection, thus resulting in ‘normal’ cortisol values. Following the switch to oral cortisone, cortisol levels before therapy were low, thus confirming post-surgical hypocortisolism. Hypocortisolism remained throughout the first year after surgery, and glucocorticoid therapy was continued. Sixteen months after surgery, baseline cortisol levels returned to the normal range; cortisone therapy was therefore tapered and a further hormonal check was scheduled. Assessment of the cortisol response to ACTH stimulation testing would be helpful in order to check the resumption of the residual adrenal function.
A peculiar aspect of our case was the occurrence of a psycho-organic syndrome together with the finding of a splenial lesion on brain imaging, which was deemed secondary to metabolic injury. Indeed, the increased cortisol levels present in patients with Cushing’s disease are detrimental to the white matter of the brain, including the corpus collosum, causing subsequent clinical derangements (22).
Besides the direct effects of hypercortisolism, the splenial damage was also probably due to long-standing hypertension, worsened by newly occurring catecholamine hypersecretion and diabetes. Together with the normalization of cortisol and glycaemic levels, and of BP, a partial reduction in the splenial damage was observed on two subsequent MR examinations, and the patient's neurological condition slowly improved until she fully recovered.
In our patient, thorough germinal genetic testing for the commonest pheochromocytoma/paraganglioma (PPGL) genes proved negative. Since approximately 40% of these tumours have germline mutations, genetic testing is recommended regardless of the patient’s age and family history. In the absence of syndromic, familial or metastatic presentation, the selection of genes for testing may be guided by the tumour location and biochemical phenotype.
Alterations of the PPGL genes can be divided into two groups: 10 genes (RET, VHL, NF1, SDHD, SDHAF2, SDHC, SDHB, SDHA, TMEM127 and MAX) that have well-defined genotype–phenotype correlations, thus allowing to tailor imaging procedures and medical management, and a group of other emerging genes, which lack established genotype–phenotype associations; for patients in whom mutations of genes belonging to this second group are detected, and hence hereditary predisposition is established, only general medical surveillance and family screening can be planned (23, 24).
In conclusion, our case highlights the importance of investigating patients with hypertension and metabolic derangements such as diabetes and hypokalaemia, since these findings may be a sign of newly occurring EAS, which, in rare cases, may be due to an ACTH-secreting pheochromocytoma. Since the additive effect of cortisol and catecholamine can cause dramatic clinical consequences, the possibility of an ACTH-secreting pheochromocytoma should be taken into account in the presence of an adrenal mass. EAS must be considered an endocrine emergency requiring urgent multi-specialist treatment. Surgery, whenever possible, is usually curative, and anatomic brain damage, as ascertained in our patient, may be at least partially reversible.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This study did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector. The study was approved by the Local Ethics Committee (no: 732/2022).
Patient consent
The patient provided written informed consent.
Author contribution statement
All authors contributed equally to the conception, writing and editing of the manuscript. L Foppiani took care of the patient during hospitalization and in the outpatient department, performed the metabolic and endocrine work-up, conceived the study, analysed the data and wrote the manuscript. MG Poeta evaluated the patient during hospitalization with regard to neurological problems and planned the related work-up (brain imaging procedures and rachicentesis). M Rutigliani analysed the histological specimens and performed immunohistochemical studies. S Parodi performed CT and MR scans and analysed the related images. U Catrambone performed the left adrenalectomy. L Cavalleri performed general anaesthesia and assisted the patient during the surgical and post-surgical periods. G Antonucci revised the manuscript. P Del Monte helped in the endocrine work-up, in the evaluation of hormonal data and in the revision of the manuscript. A Piccardo performed 18F-FDG PET/CT and analysed the related images.
Acknowledgement
The work of Prof Silvia Morbelli in performing and analysing 68Ga-DOTATOC PET/CT is gratefully acknowledged.
References
-
1↑
Pivonello R, Isidori AM, De Martino MC, Newell-Price J, Biller BMK, Colao A. Complications of Cushing's syndrome: state of the art. The Lancet Diabetes & Endocrinology 2016 4 611–629. (https://doi.org/10.1016/S2213-8587(1600086-3)
-
2↑
Fleseriu M, Auchus R, Bancos I, Ben-Shlomo A, Bertherat J, Biermasz NR, Boguszewski CL, Bronstein MD, Buchfelder M, Carmichael JD, et al.Consensus on diagnosis and management of Cushing's disease: a guideline update. Lancet. Diabetes and Endocrinology 2021 9 847–875. (https://doi.org/10.1016/S2213-8587(2100235-7)
-
3↑
Gabi JN, Milhem MM, Tovar YE, Karem ES, Gabi AY, & Khthir RA. Severe Cushing syndrome due to an ACTH-producing pheochromocytoma: a case presentation and review of the literature. Journal of the Endocrine Society 2018 2 621–630. (https://doi.org/10.1210/js.2018-00086)
-
4↑
Young J, Haissaguerre M, Viera-Pinto O, Chabre O, Baudin E, & Tabarin A. Management of endocrine disease: Cushing's syndrome due to ectopic ACTH secretion: an expert operational opinion. European Journal of Endocrinology 2020 182 R29–R58. (https://doi.org/10.1530/EJE-19-0877)
-
5↑
Hayes AR, & Grossman AB. The ectopic adrenocorticotropic hormone syndrome: rarely easy, always challenging. Endocrinology and Metabolism Clinics of North America 2018 47 409–425. (https://doi.org/10.1016/j.ecl.2018.01.005)
-
6↑
Hayes AR, & Grossman AB. Distinguishing Cushing's disease from the ectopic ACTH syndrome: needles in a haystack or hiding in plain sight? Journal of Neuroendocrinology 2022 34 e13137. (https://doi.org/10.1111/jne.13137)
-
7↑
Sakuma I, Higuchi S, Fujimoto M, Takiguchi T, Nakayama A, Tamura A, Kohno T, Komai E, Akina Shiga A, Nagano H, et al.Cushing syndrome due to ACTH-secreting pheochromocytoma, aggravated by glucocorticoid-driven positive-feedback loop. Journal of Clinical Endocrinology and Metabolism 2016 101 841–846. (https://doi.org/10.1210/jc.2015-2855)
-
8↑
Barrett C, van Uum SH, & Lender JW. Risk of catecholaminergic crises following glucocorticoid administration in patients with an adrenal mass: a literature review. Clinical Endocrinology 2015 83 622–628. (https://doi.org/10.1111/cen.12813)
-
9↑
Ballav C, Naziat A, Mihai R, Karavitaki N, Ansorge O, & Grossman AB. Mini-review: pheochromocytomas causing the ectopic ACTH syndrome. Endocrine 2012 42 69–73. (https://doi.org/10.1007/s12020-012-9646-7)
-
10↑
Elliott PF, Berhane T, Ragnarsson O, & Falhammar H. Ectopic ACTH- and/or CRH-Producing pheochromocytomas. Journal of Clinical Endocrinology and Metabolism 2021 106 598–608. (https://doi.org/10.1210/clinem/dgaa488)
-
11↑
Isidori AM, Kaltsas GA, Pozza C, Frajese V, Newell-Price J, Reznek RH, Jenkins PJ, Monson JP, Grossman AB, & Besser GM. The ectopic adrenocorticotropin syndrome: clinical features, diagnosis, management, and long-term follow-up. Journal of Clinical Endocrinology and Metabolism 2006 91 371–377.. (https://doi.org/10.1210/jc.2005-1542)
-
12↑
Salgado LR, Fragoso MCB, Knoepfelmacher M, Machado MC, Domenice S, Pereira MA, & de Mendonça BB. Ectopic ACTH syndrome: our experience with 25 cases. European Journal of Endocrinology 2006 155 725–733. (https://doi.org/10.1530/eje.1.02278)
-
13↑
Paleń-Tytko JE, Przybylik-Mazurek EM, Rzepka EJ, Pach DM, Sowa-Staszczak AS, Gilis-Januszewska A, & Hubalewska-Dydejczyk AB. Ectopic ACTH syndrome of different origin: diagnostic approach and clinical outcome. Experience of one clinical centre. PLoS One 2020 15 e0242679. (https://doi.org/10.1371/journal.pone.0242679)
-
14↑
Krumeich LN, Cucchiara AJ, Nathanson KL, Kelz RR, Fishbein L, Fraker DL, Roses RE, Cohen DL, & Wachtel H. Correlation between plasma catecholamines, weight, and diabetes in pheochromocytoma and paraganglioma. Journal of Clinical Endocrinology and Metabolism 2021 106 e4028–e4038. (https://doi.org/10.1210/clinem/dgab401)
-
15↑
Bihain F, Nomine-Criqui C, Guerci P, Gasman S, Klein M, & Brunaud L. Management of patients with treatment of pheochromocytoma: a critical appraisal. Cancers (Basel) 2022 14 3845. (https://doi.org/10.3390/cancers14163845)
-
16↑
Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, & Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: 20 years’ experience at the National Institute of Health. Journal of Clinical Endocrinology and Metabolism 2005 90 4955–4962. (https://doi.org/10.1210/jc.2004-2527)
-
17↑
Varlamov E, Hinojosa-Amaya JM, Stack M, & Fleseriu M. Diagnostic utility of gallium-68-somatostatin receptor PET/CT in ectopic ACTH-secreting tumours: a systematic literature review and single-center clinical experience. Pituitary 2019 22 445–455. (https://doi.org/10.1007/s11102-019-00972-w)
-
18↑
Varlamov EV, Han AJ, & Fleseriu M. Updates in adrenal steroidogenesis inhibitors for Cushing's syndrome. A practical guide. Best Practice and Research. Clinical Endocrinology and Metabolism 2021 35 101490. (https://doi.org/10.1016/j.beem.2021.101490)
-
19↑
Marques JVO, & Boguszewski CL. Medical therapy in severe hypercortisolism. Best Practice and Research. Clinical Endocrinology and Metabolism 2021 35 101487. (https://doi.org/10.1016/j.beem.2021.101487)
-
20↑
Sharma ST, & Nieman LK. Prolonged remission after long-term treatment with steroidogenesis inhibitors in Cushing's syndrome caused by ectopic ACTH secretion. European Journal of Endocrinology 2012 166 531–536. (https://doi.org/10.1530/EJE-11-0949)
-
21↑
Martinez AD, Feelders RA, de Herder WW, Castaño JP, Moreno MAG, Dogan F, van Dungen R, van Koetsveld P, & Hofland LJ. Effects of ketoconazole on ACTH-producing and non-ACTH-producing neuroendocrine tumor cells. Hormones and Cancer 2019 107–119. (https://doi.org/10.1007/s12672-019-00361-6)
-
22↑
Cui M, Zhou T, Feng S, Liu X, Wang F, Zhang Y, & Yu X. Altered microstructural pattern of white matter in Cushing’s disease identified by automated fiber quantification. NeuroImage. Clinical 2021 31 102770. (https://doi.org/10.1016/j.nicl.2021.102770)
-
23↑
Lenders JWM, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SKG, Hassan Murad MH, Naruse M, Pacak K, Young WF Jr & Endocrine Society. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. Journal of Clinical Endocrinology and Metabolism 2014 99 1915–1942. (https://doi.org/10.1210/jc.2014-1498)
-
24↑
Sarkadi B, Saskoi E, Butz H, & Patocs A. Genetics of pheochromocytomas and paragangliomas determine the therapeutical approach. International Journal of Molecular Sciences 2022 23 1450. (https://doi.org/10.3390/ijms23031450)
From https://edm.bioscientifica.com/view/journals/edm/2023/2/EDM22-0308.xml
-
1
-
-
Introduction: The first-line treatment for Cushing’s disease is transsphenoidal surgery for pituitary tumor resection. Ketoconazole has been used as a second-line drug despite limited data on its safety and efficacy for this purpose. The objective of this meta-analysis was to analyze hypercortisolism control in patients who used ketoconazole as a second-line treatment after transsphenoidal surgery, in addition to other clinical and laboratory criteria that could be related to therapeutic response.
Methods: We searched for articles that evaluated ketoconazole use in Cushing’s disease after transsphenoidal surgery. The search strategies were applied to MEDLINE, EMBASE, and SciELO. Independent reviewers assessed study eligibility and quality and extracted data on hypercortisolism control and related variables such as therapeutic dose, time, and urinary cortisol levels.
Results: After applying the exclusion criteria, 10 articles (one prospective and nine retrospective studies, totaling 270 patients) were included for complete data analysis. We found no publication bias regarding reported biochemical control or no biochemical control (p = 0.06 and p = 0.42 respectively). Of 270 patients, biochemical control of hypercortisolism occurred in 151 (63%, 95% CI 50-74%) and no biochemical control occurred in 61 (20%, 95% CI 10-35%). According to the meta-regression, neither the final dose, treatment duration, nor initial serum cortisol levels were associated with biochemical control of hypercortisolism.
Conclusion: Ketoconazole can be considered a safe and efficacious option for Cushing’s disease treatment after pituitary surgery.
Systematic review registration: https://www.crd.york.ac.uk/prospero/#searchadvanced, (CRD42022308041).
1 Introduction
Cushing’s disease (CD) results from an adrenocorticotropic hormone (ACTH) secreting pituitary tumor, which leads to chronic hypercortisolism (1, 2). It is a potentially fatal disease, with mortality rates up to 3.7 times higher than the general population (3, 4). CD is three times more common in women.
According to consensus, the first-line treatment for CD is pituitary tumor resection surgery with the transsphenoidal technique (4, 5), which achieves short-term biochemical control rates of 60 to 80%, depending on the experience of the treatment center. In long-term follow-up, recurrence rates range from 20 to 30% even in cases with complete initial biochemical control (6, 7).
Medication is a therapeutic option in patients who do not achieve biochemical control with transsphenoidal surgery (TSS), have recurrent hypercortisolism, and have contraindications or high surgical risk, or it can be used while waiting for the efficacy of radiation techniques (8). In such cases, adrenal-blocking drugs become important.
Ketoconazole is an antifungal drug, a synthetic imidazole derivative that blocks multiple enzymes involved in adrenal steroidogenesis pathways (CYP11A1, CYPP17, CYP11B2, and CYP11B1). It was recently approved for use in CD by the European Union (9) and has been recommended for off-label use in the United States (2, 10, 11). Although recommended by professional guidelines (not regulatory authorities) for hypercortisolism, its use as an antifungal has been more restricted since regulatory agencies in Europe and the United States have issued statements regarding its high risk of hepatotoxicity, including reported deaths from liver failure (12, 13). Recently, a levorotatory derivative (Levoketoconazole) with estimated lower hepatotoxicity was introduced (14).
Clinical studies evaluating the efficacy and adverse effects of ketoconazole in CD are scarce. Their limited and heterogeneous samples include hypercortisolism control as a first-line therapy or after TSS and they include patients with ACTH-dependent Cushing’s syndrome with indeterminate etiology (11–13).
Two recent meta-analyses had divergent results regarding hypercortisolism remission rates with ketoconazole use: 46% vs. 64% (15, 16). Adverse effects, treatment interruption, and treatment-associated deaths have also been reported. Thus, studies evaluating the efficacy of ketoconazole for its main indication and continued or recurrent hypercortisolism after TSS are not currently available.
This meta-analysis aimed to analyze the prevalence of biochemical control of hypercortisolism in CD patients who used ketoconazole as a second-line therapy after TSS, in addition to clinical and laboratory parameters that can predict therapeutic response and serious adverse effects due to ketoconazole treatment.
2 Materials and methods
This systematic review and meta-analysis study was performed according to the PRISMA system (17) and was registered in the International Prospective Register of Systematic Reviews (CRD42022308041).
2.1 Identification of studies
A search was performed in three databases: MEDLINE, EMBASE, and SciELO. In MEDLINE, using the Medical Subject Headings “Pituitary ACTH hypersecretion” or “Cushing’s disease” and “Ketoconazole” or “Fluconazole”, 305 articles were found. In EMBASE, using the Emtree terms “Cushing’s disease” and “ketoconazole” or “fluconazole”, 544 results were found. In SciELO, using the terms “Cushing’s disease” and “Ketoconazole” or “fluconazole”, five articles were found.
The complete search strategy can be found in Supplementary Material 1. The searches were performed in June 2021 and updated in May 2022 although no new studies were added to the analysis through this step. A manual search was performed for references to reviews and meta-analyses in the included studies, as well as systematic reviews or articles on related topics. Every potential article was considered eligible for review, with no language limitations. Whenever necessary, authors were contacted to confirm information or supply missing data.
2.2 Selection criteria
We selected observational, case-control, or clinical trials that included CD patients diagnosed through clinical manifestations in association with at least two positive screenings for hypercortisolism, baseline ACTH > 20 pg/ml, pituitary adenoma confirmed in surgery, bilateral petrosal sinus catheterization, or pituitary MRI showing a lesion > 6 mm (18). Patients must have undergone transsphenoidal surgery as first-line therapy, either without postoperative remission or with recurrence during clinical follow-up. Consequently, ketoconazole was used as a second-line treatment to control hypercortisolism. Studies of patients who received radiotherapy concomitantly with ketoconazole were not excluded.
2.3 Study selection, data extraction, and quality assessment
Two authors (CV and ACVM) performed independent searches in the databases, selecting potential studies based on titles and abstracts for further analysis of the complete articles. Inter-rater agreement was 0.88 according to Cohen’s kappa coefficient (95% CI, 0.83-0.93) for the selected studies. Disagreements were resolved by consensus between the investigators (CV and ACVM) or when necessary, by a discussion with a third investigator (MAC). Baseline characteristics and outcomes were extracted from studies that met the inclusion criteria, including baseline and post-drug cortisol measurements, mean and maximum treatment duration, ketoconazole dose, potential adverse effects, and drug intolerance. The considered outcomes were the prevalence of complete, partial (reduction of > 50% in cortisol levels despite incomplete normalization of 24-h UFC), or no biochemical control of hypercortisolism with ketoconazole use.
Data were extracted only when the studies reported ketoconazole use after transsphenoidal surgery (TSS). Studies that did not subdivide ketoconazole data into pre-and post-transsphenoidal surgery were excluded.
Disagreements about data extraction were discussed until a consensus was reached. The original authors were contacted by e-mail to resolve questions or obtain missing data. Study quality was evaluated using a modified Newcastle–Ottawa scale (19).
2.4 Data analysis
Rates of complete, partial, and no biochemical control were analyzed across all included studies and the pooled prevalence was calculated. Cochrane’s χ2 and I² tests were used to assess heterogeneity between studies, and p = 0.05 was considered significant. Incidence estimates were obtained by random effects models. Meta-regression was performed to analyze the relationship between ketoconazole dose, treatment time, and baseline cortisol level.
Publication bias was assessed with a funnel plot that assesses the incidences in relation to the standard error of each study, which was determined using the Begg and Egger tests. Meta-analysis was performed using R version 4.1.2 and R META package version 4.19.2.
3 Results
Electronic and manual database searches resulted in 735 studies, of which 652 were excluded after analyzing the titles and abstracts. We selected 83 studies for full-text review. After applying the exclusion criteria, 10 articles remained (totaling 270 patients) for analysis and complete data extraction (10, 20–28). The flow diagram is shown in Figure 1. No articles using the term fluconazole in the context of CD were found in the searches.
Figure 1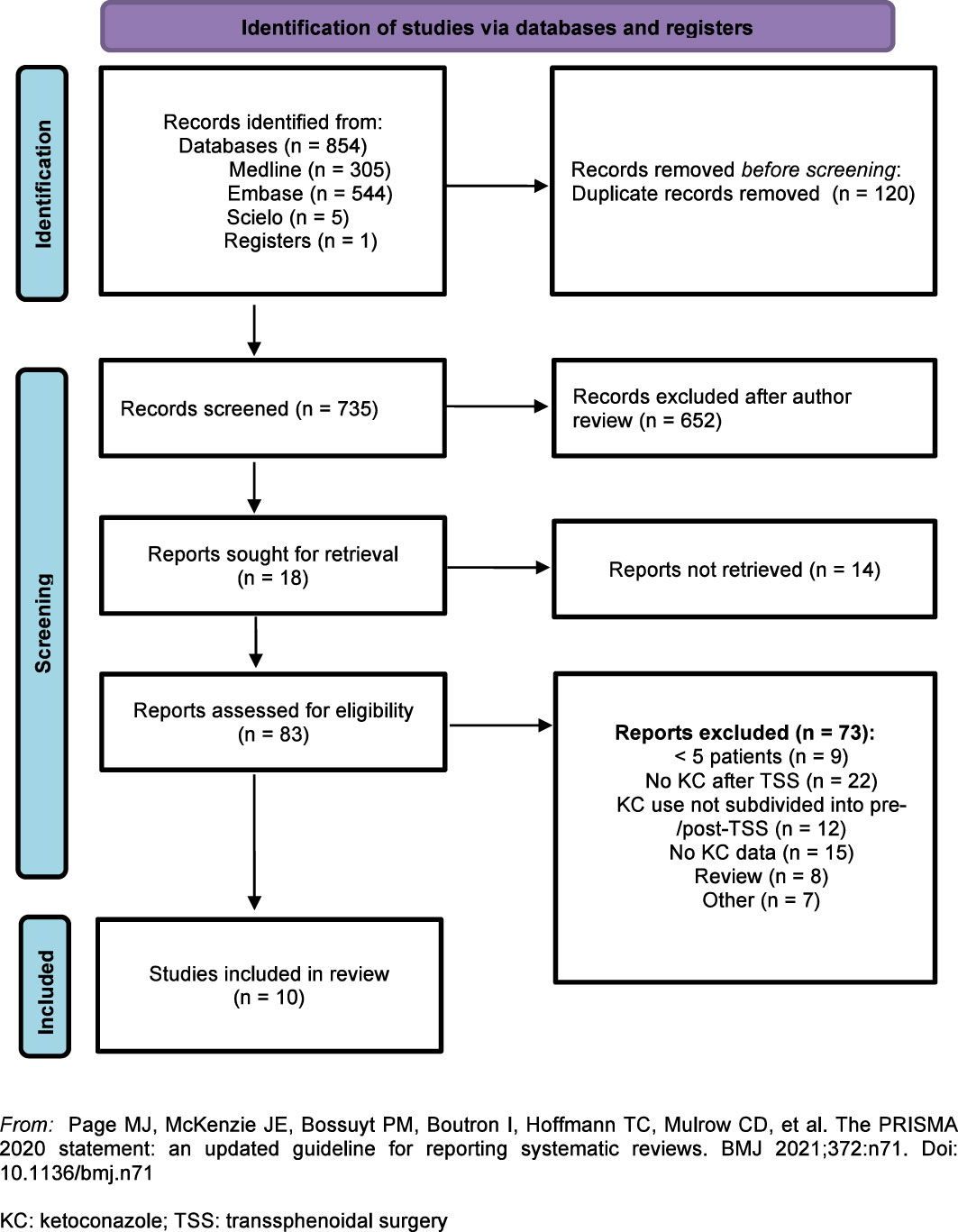
Figure 1 Flow diagram: Identification and selection of articles for the meta-analysis.
All of the selected studies used normalized 24-h UFC levels as a criterion for biochemical control of hypercortisolism except for one (24), which used serum cortisol level and the suppression test with 2 mg of dexamethasone (Liddle test).
Most patients were women and were treated with ketoconazole for a mean of 31.4 months and a maximum of 45 months. Details of each included study are presented in Table 1. Unpublished data from a conference abstract from a Brazilian cohort were included and were supplemented through direct contact with the authors (27).
Table 1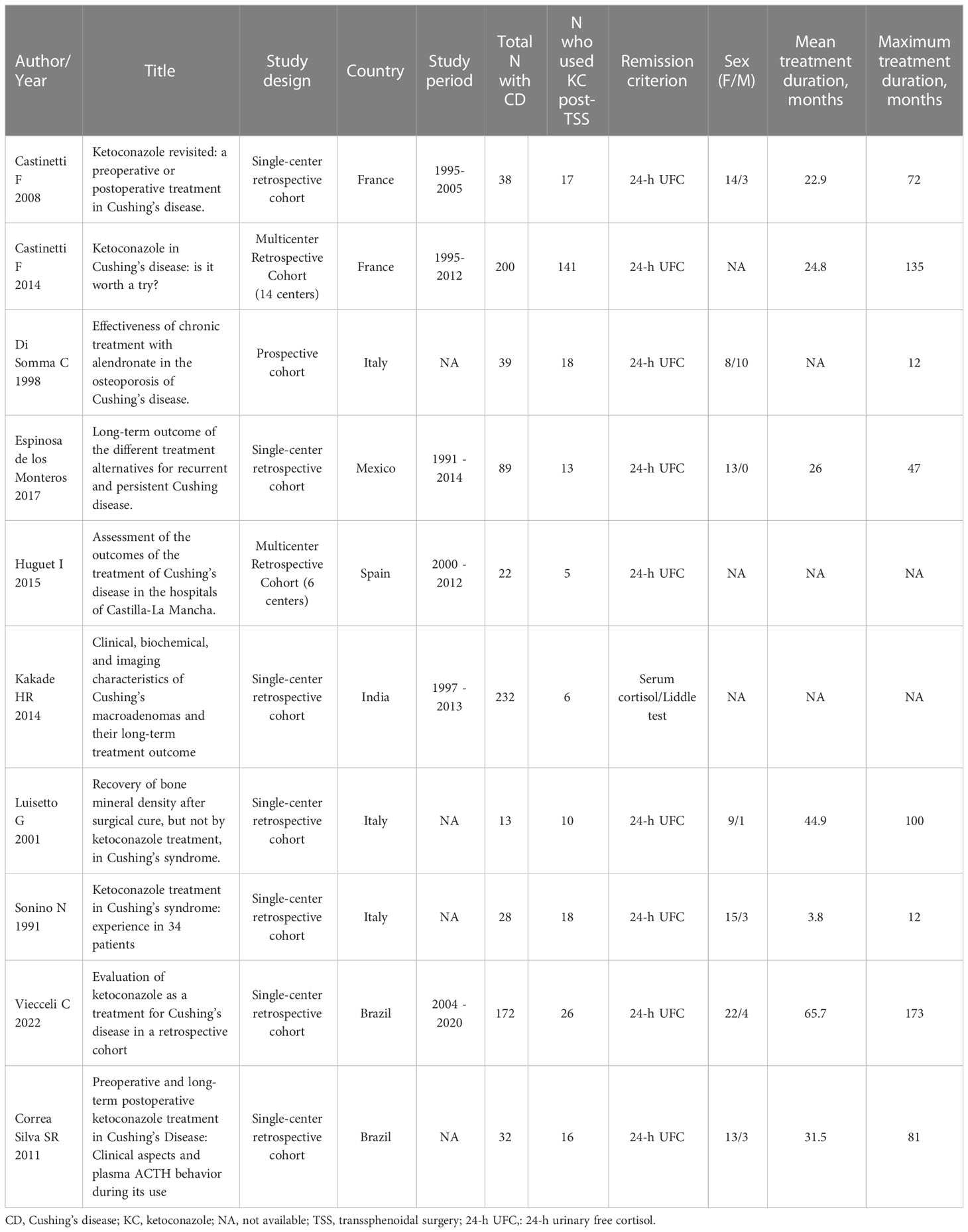
Table 1 Characteristics of the included studies.
The study quality analysis is shown in Table 2. In general, the quality of the articles was adequate. Some data could not be extracted due to uncertainty about when TSS had been performed and ketoconazole therapy had begun. In such cases, the authors were contacted and, if they did not respond by the time of the analyses, the data were excluded. The study by Huguet et al. (23) was excluded from the analysis of the “no biochemical control” variable for not mentioning non-remission as a possible outcome.
Table 2
Table 2 Quality of the included studies (one-star maximum for each item, except comparability of cohorts, with two maximum).
Begg and Egger’s tests were performed to assess publication bias regarding biochemical control of hypercortisolism. Since the results were not significant, there was no need to perform a trim-and-fill analysis. Funnel Plots (Figures 2, 3) demonstrate the lack of publication bias regarding biochemical control and no biochemical control (p = 0.06 and p = 0.42, respectively).
Figure 2
Figure 2 Funnel Plot of hypercortisolism remission with Ketoconazole.
Figure 3
Figure 3 Funnel Plot of hypercortisolism non-remission with ketoconazole.
3.1 Control of hypercortisolism (biochemical control)
Ten studies (270 patients) indicated the prevalence of biochemical control of hypercortisolism in patients who underwent TSS and received ketoconazole as a second-line therapy. A total of 151 patients had complete biochemical control (63%; 95% CI, 50-74%; see Figure 4). We performed a meta-analysis without including Correa Silva’s unpublished data, and the prevalence of hypercortisolism remission remained at 63%. These charts can be found in the Supplementary Material.
Figure 4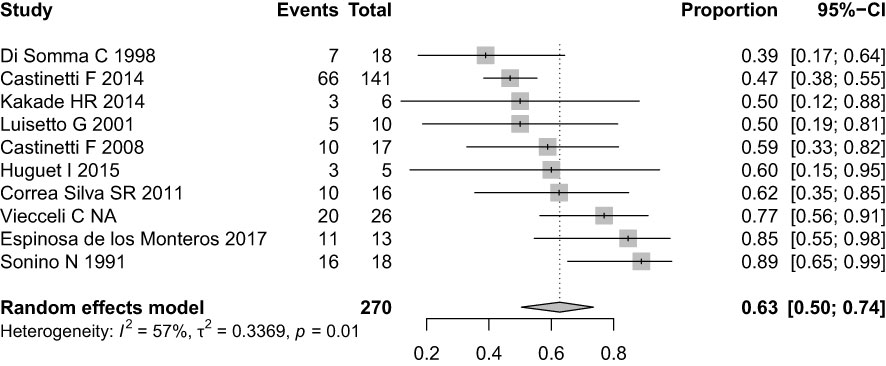
Figure 4 Forest plot of hypercortisolism remission with Ketoconazole.
The high variability between studies is partly explained by the clinical differences between cohorts, which explain the 39 to 89% variation in remission rates. The lowest complete remission rate, 39%, was found in Di Somma et al. However, in addition to being the only prospective study, there was a high rate of partial biochemical control (61%), and no patient was classified as no biochemical control. This cohort also had the highest mean baseline cortisol levels (1413 nmol/24h, 9.46 times above the upper reference limit) and the lowest mean final ketoconazole dose (400 mg daily). The highest remission rate, 89%, was found in Sonino et al., a retrospective cohort, which might explain why ketoconazole was administered only in patients with a more favorable clinical response. Heterogeneity was 57% in this analysis.
No biochemical control occurred in 61 of 270 patients or 20% of the sample (95% CI, 10-35%) (Figure 5). The four cohorts with the highest rates of non-remission, Kakade HR et al. (50%), Luisetto G et al. (50%), Castinetti F et al. (41%), and Espinosa de los Monteros et al. (26.7%) did not involve the concept of partial biochemical control, which was used in the other cohorts. Heterogeneity was 4% in this analysis.
Figure 5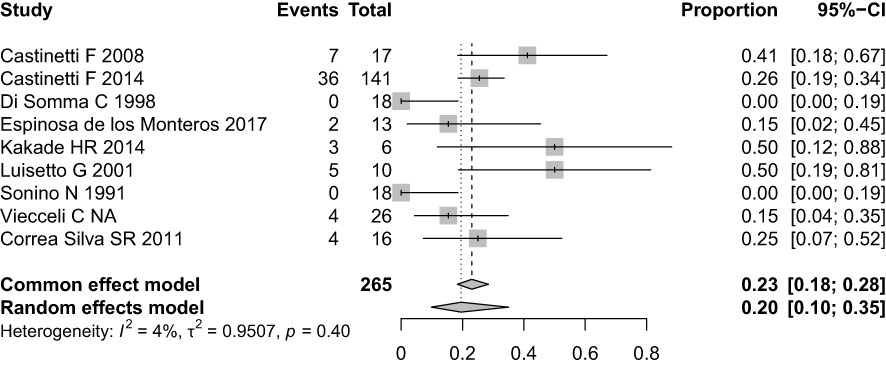
Figure 5 Forest plot of hypercortisolism non-remission with ketoconazole.
Although the concept of partial response was not addressed directly in most studies, some patients experienced a reduction of > 50% in cortisol levels despite incomplete normalization. This condition was described in five cohorts (10, 21, 26, 27, 28), demonstrating partial benefits from ketoconazole in 59 patients (21.7%).
Only five papers mentioned how many patients underwent radiotherapy during treatment with ketoconazole; at least 59 patients (21%) received radiotherapy treatment concomitantly or subsequent to ketoconazole (10, 22, 23, 27, 28).
3.2 Adverse effects
Although all of the studies described adverse effects from ketoconazole, only two provided information about them after TSS (26, 28). The following stood out among the main adverse effects: elevated transaminase levels, diarrhea, abdominal pain, skin rash, gynecomastia, and adrenal insufficiency. Medication discontinuation due to intolerance was reported in three studies (10, 20, 28). Due to insufficient data, it was not possible to perform a meta-analysis of the prevalence of adverse effects. No deaths related to ketoconazole were reported in any study.
3.3 Meta-regression
In studies that evaluated hypercortisolism remission, meta-regression was used to analyze which variables influenced the occurrence or not of biochemical control. Both the final dose of ketoconazole (six studies with a mean dose of 628 mg/day: range 400 mg to 779 mg/day), the duration of drug treatment (seven studies with a mean duration of 31 months), and the baseline 24-h UFC levels (seven studies with a mean of 4.48 times above the reference value) showed no association with hypercortisolism remission (data not shown).
4 Discussion
Drug treatment in CD is reserved only for patients with no biochemical control after TSS, in those who are not candidates for surgical treatment, or in those awaiting the effects of radiotherapy (2, 4). The available drugs in this context act in several ways: as adrenal blockers (ketoconazole, osilodrostat, metyrapone, mitotane, levoketoconazole, and etomidate), somatostatin receptor ligands (pasireotide), dopamine receptor agonists (cabergoline), or as glucocorticoid receptor blockers (mifepristone) (2, 29). These drugs must be prescribed considering aspects such as the potential for remission, potential adverse effects, availability, and cost. Moreover, no single drug has yet been demonstrated as superior to the others (2, 30, 31).
Comparing our analyses with previous studies, we found that hypercortisolism control in patients who had already undergone TSS was higher than in studies that did not subdivide ketoconazole use into pre- and post-transsphenoidal surgery or in studies evaluating multiple etiologies of hypercortisolism (15, 16, 32).
Our meta-analysis evaluated 10 studies from different countries and ethnic groups regarding CD treatment with ketoconazole due to non-remission or recurrence after TSS. The hypercortisolism biochemical control rate we found after TSS (63%) was greater than some prospective studies evaluating current drugs such as levoketoconazole but was also similar to that found in a systematic review by Pivonello et al. (64%) (14, 32). However, it was higher than that found in the most recent meta-analysis (36 to 46%) (15). These two systematic reviews (14, 15) did not subdivide ketoconazole use into pre- and post-transsphenoidal surgery, which can significantly impact the hypercortisolism control rate. A multicenter study by Castinetti et al. showed greater efficacy in patients who had already undergone TSS (68% control) compared to preoperative use (48.7% control) (10). These findings may be due to the fact that assessing patients with different states of hypercortisolism broadens the sample beyond only CD patients (i.e., probably including patients with ectopic ACTH syndrome and other etiologies) and, thus, the percentage of controlled patients may be lower.
According to the literature, even without complete biochemical control, patients who present some reduction in serum cortisol levels, partial biochemical control, or improvement in any associated comorbidities are candidates for continuing ketoconazole alone or in a possible association with other medications (2). Our meta-analysis found that such was the case in 59 patients. Although the concept of partial response was not addressed directly in most of the included studies, some individuals experienced a > 50% reduction in cortisol levels but not complete normalization. By analyzing the overall rate of non-responders (20%), we can extrapolate that approximately 80% of patients treated with ketoconazole experienced some improvement in cortisol levels, which in itself demonstrates the medication’s efficacy.
Although we considered the hypercortisolism biochemical control rate to be satisfactory with ketoconazole, many patients may lose biochemical control over the course of treatment or have long-term oscillations, and it has been suggested that this can occur in up to 23% of those who achieved initial control using the drug (2, 32), which shows the dynamic nature of their treatment and the constant challenge in clinical practice. This could not be established in our meta-analysis due to the lack of reported data (15, 16, 32). Although tumor size is not necessarily related to cortisol levels in CD, those with macroadenomas have a lower chance of remission after TSS (2, 33). Patients who use ketoconazole preoperatively may already have larger lesions, which makes surgery difficult, or active pituitary lesions, which can reduce the ability to achieve control through medication. In our meta-analysis, only two studies described tumor size and correlated it with remission after ketoconazole therapy (10, 24).
The hypothesis that patients with lower pre-treatment serum cortisol levels or who used higher doses of ketoconazole would have higher biochemical control rates was not confirmed since we found no relationship between longer duration of use and higher remission rates. The data included in this review do not provide a profile of patients most likely to benefit from ketoconazole treatment. Other reviews of ketoconazole therapy in any context of Cushing’s syndrome have found that up to 20% of patients experience adverse effects such as elevated transaminase levels, with the majority being asymptomatic moderate elevation, i.e., < 5 times the upper limit of normality. These hepatic changes do not appear dose-dependent and are usually reversed within 2 to 12 weeks after ketoconazole discontinuation or dose reduction (34). When compared, up to 32% of participants experienced mild adverse effects in the levoketoconazole study, with 13% having to discontinue treatment (14). Our analyses have several limitations since nine of the 10 primary studies that were included in the meta-analysis were retrospective and uncontrolled in design. We could find no randomized clinical trials, and we know that only randomized, controlled trials with an intention to treat analysis can provide accurate estimates of drug efficacy. New therapeutic options are under investigation in clinical trials and will likely bring more robust data about hypercortisolism control in CD.
Despite the limitations, consensus continues to indicate adrenal blockers, including ketoconazole, for patients with moderate CD and no visible lesions in MRI. The recommendation is that drug therapy should be individualized, based on the patient’s clinical picture, hypercortisolism severity, and medication availability and cost, so that treatment is optimized and applied for the necessary period of time (2, 33, 35, 36).
5 Conclusion
Our meta-analysis showed that ketoconazole effectively controlled hypercortisolism in approximately 63% of CD patients when used according to its principal indication, i.e., in patients without remission after TSS. No association was found between hypercortisolism biochemical control and total medication dose, treatment duration, or initial serum cortisol levels. No serious adverse effects or treatment-related deaths were observed in these patients. These findings indicate that based on the current literature available, ketoconazole is an efficacious and safe drug for treating active CD after pituitary surgery.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
CV, SPG and MAC created the research format. CV and ACVM developed the search strategies and independently applied the eligibility criteria, subsequently extracting the data. CV and ACVM performed a peer review of the data and assessed risk of bias. CV and VNH performed the meta-analysis. MAC oversaw all phases of the meta-analysis and arbitrated conflicts of opinion. SPG and TCR participated in the final data review and discussion. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the “Coordena̧cão de Aperfei̧coamento de Pessoal de Ńıvel Superior” (CAPES), Ministry of Health - Brazil, through a PhD scholarship; and the Research Incentive Fund (FIPE) of Hospital de Cĺınicas de Porto Alegre (HCPA) and Programa de Excelência Acadêmica from CAPES (PROEX).
Acknowledgments
The authors would like to thank Ana Cabral, librarian at the Federal University of Rio Grande do Sul, for her availability and assistance with the database searches and Professor Silvia Regina Correa da Silva for kindly providing additional unpublished data from her study.
Conflict of interest
TCR received a CNPQ research grant. MAC worked on clinical research for Crinetics and on the advisory board for Novo Nordisk.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1145775/full#supplementary-material
References
1. Fleseriu M, Castinetti F. Updates on the role of adrenal steroidogenesis inhibitors in cushing’s syndrome: a focus on novel therapies. Pituitary (2016). 643–53. doi: 10.1007/s11102-016-0742-1
2. Fleseriu M, Auchus R, Bancos I, Ben-Shlomo A, Bertherat J, Biermasz NR, et al. Consensus on diagnosis and management of cushing’s disease: a guideline update. Lancet Diabetes Endocrinol (2021) 9(12):847–75. doi: 10.1016/S2213-8587(21)00235-7
3. Nieman LK, Biller BMK, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The diagnosis of cushing’s syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab (2008) 93(5):1526–40. doi: 10.1210/jc.2008-0125
4. Nieman LK, Biller BMK, Findling JW, Murad MH, Newell-Price J, Savage MO, et al. Treatment of cushing’s syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab (2015) 100(8):2807–31. doi: 10.1210/jc.2015-1818
5. Clayton RN, Raskauskiene D, Reulen RC, Jones PW. Mortality and morbidity in cushing’s disease over 50 years in Stoke-on-Trent, UK: audit and meta-analysis of literature. J Clin Endocrinol Metab (2011). 632–42. doi: 10.1210/jc.2010-1942
6. Patil CG, Prevedello DM, Lad SP, Vance ML, Thorner MO, Katznelson L, et al. Late recurrences of cushing’s disease after initial successful transsphenoidal surgery. J Clin Endocrinol Metab (2008) 93(2):358–62. doi: 10.1210/jc.2007-2013
7. Hofmann BM, Hlavac M, Martinez R, Buchfelder M, Müller OA, Fahlbusch R. Long-term results after microsurgery for cushing disease: experience with 426 primary operations over 35 years. J Neurosurg (2008) 108(1):9–18. doi: 10.3171/JNS/2008/108/01/0009
8. Rubinstein G, Osswald A, Zopp S, Ritzel K, Theodoropoulou M, Beuschlein F, et al. Therapeutic options after surgical failure in cushing’s disease: a critical review. Best Pract Res Clin Endocrinol Metab (2019) 33(2):101270. doi: 10.1016/j.beem.2019.04.004
9. Agency EM. Ketoconazole HRA recommended for approval in cushing’s syndrome European medicines agency facilitates patients’ access to a treatment of a. (2014) 44(September).
10. Castinetti F, Guignat L, Giraud P, Muller M, Kamenicky P, Drui D, et al. Ketoconazole in cushing’s disease: is it worth a try. J Clin Endocrinol Metab (2014) 99(5):1623–30. doi: 10.1210/jc.2013-3628
11. Castinetti F, Nieman LK, Reincke M, Newell-Price J. Approach to the patient treated with steroidogenesis inhibitors. J Clin Endocrinol Metab (2021) 106(7):2114–23. doi: 10.1210/clinem/dgab122
12. Greenblatt DJ, Mikus G. Ketoconazole and liver injury: a five-year update. Clin Pharmacol Drug Dev (2019) 8(1):6–8. doi: 10.1002/cpdd.652
13. Yan JY, Nie XL, Tao QM, Zhan SY, Zhang Y. Ketoconazole associated hepatotoxicity: a systematic review and meta-analysis. BioMed Environ Sci (2013) 26(7):605–10. doi: 10.3967/0895-3988.2013.07.013
14. Fleseriu M, Pivonello R, Elenkova A, Salvatori R, Auchus RJ, Feelders RA, et al. Efficacy and safety of levoketoconazole in the treatment of endogenous cushing’s syndrome (SONICS): a phase 3, multicentre, open-label, single-arm trial. Lancet Diabetes Endocrinol (2019) 7(11):855–65. doi: 10.1016/S2213-8587(19)30313-4
15. Simões Corrêa Galendi J, Correa Neto ANS, Demetres M, Boguszewski CL, Nogueira V dos SN. Effectiveness of medical treatment of cushing’s disease: a systematic review and meta-analysis. Front Endocrinol (Lausanne) (2021) 12(September):1–12. doi: 10.3389/fendo.2021.732240
16. Broersen LHA, Jha M, Biermasz NR, Pereira AM, Dekkers OM. Effectiveness of medical treatment for cushing’s syndrome: a systematic review and meta-analysis. Pituitary (2018) 21(6):631–41. doi: 10.1007/s11102-018-0897-z
17. Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ (2021) 89:372. doi: 10.1136/bmj.n71
18. Boscaro M, Arnaldi G. Approach to the patient with possible cushing’s syndrome. J Clin Endocrinol Metab (2009) 94(9):3121–31. doi: 10.1210/jc.2009-0612
19. Wells G, Shea B OD. The Newcastle–Ottawa scale (NOS) for assessing the quality of nonrandomised studies in meta-analyses. Ottawa Hosp Res Inst (2016).
20. Castinetti F, Morange I, Jaquet P, Conte-Devolx B, Brue T. Ketoconazole revisited: a preoperative or postoperative treatment in cushing’s disease. Eur J Endocrinol (2008) 158(1):91–9. doi: 10.1530/EJE-07-0514
21. Di Somma C, Colao A, Pivonello R, Klain M, Faggiano A, Tripodi FS, et al. Effectiveness of chronic treatment with alendronate in the osteoporosis of cushing’s disease. Clin Endocrinol (Oxf) (1998) 48(5):655–62. doi: 10.1046/j.1365-2265.1998.00486.x
22. Espinosa-De-Los-Monteros AL, Sosa-Eroza E, Espinosa E, Mendoza V, Arreola R, Mercado M. Long-term outcome of the different treatment alternatives for recurrent and persistent cushing disease. Endocr Pract (2017) 23(7):759–67. doi: 10.4158/EP171756.OR
23. Huguet I, Aguirre M, Vicente A, Alramadan M, Quiroga I, Silva J, et al. Análisis de los resultados del tratamiento de la enfermedad de cushing en los hospitales de castilla-la mancha. Endocrinol y Nutr (2015) 62(5):217–23. doi: 10.1016/j.endonu.2015.02.007
24. Kakade HR, Kasaliwal R, Khadilkar KS, Jadhav S, Bukan A, Khare S, et al. Clinical, biochemical and imaging characteristics of cushing’s macroadenomas and their long-term treatment outcome. Clin Endocrinol (Oxf) (2014) 81(3):336–42. doi: 10.1111/cen.12442
25. Luisetto G, Zangari M, Camozzi V, Boscaro M, Sonino N, Fallo F. Recovery of bone mineral density after surgical cure, but not by ketoconazole treatment, in cushing’s syndrome. Osteoporos Int (2001) 12(11):956–60. doi: 10.1007/s001980170025
26. Sonino N, Boscaro M, Paoletta A, Mantero F, Zillotto D. Ketoconazole treatment in cushing’s syndrome: experience in 34 patients. Clin Endocrinol (Oxf) (1991) 35(4):347–52. doi: 10.1111/j.1365-2265.1991.tb03547.x
27. Correa-Silva SR, Gaeta P, Alves GM, Alves Martins MR, Abucham J, Judith Lengyel A-M. Preoperative and long-term postoperative ketoconazole treatment in cushing disease: clinical aspects and plasma acth behavior during its use. Endocr Rev (2011) 32(3).
28. Viecceli C, Mattos ACV, Costa MCB, de Melo RB, Rodrigues TdaC, Czepielewski MA. Evaluation of ketoconazole as a treatment for cushing’s disease in a retrospective cohort. Front Endocrinol (Lausanne) (2022) 13(October). doi: 10.3389/fendo.2022.1017331
29. Gadelha MR, Neto LV. Efficacy of medical treatment in cushing’s disease: a systematic review. Clin Endocrinol (Oxf) (2014) 80(1):1–12. doi: 10.1111/cen.12345
30. Fleseriu M, Petersenn S. New avenues in the medical treatment of cushing’s disease: corticotroph tumor targeted therapy. J Neurooncol (2013) 114(1):1–11. doi: 10.1007/s11060-013-1151-1
31. Fleseriu M, Petersenn S. Medical management of cushing’s disease: what is the future? Pituitary (2012) 15(3):330–41. doi: 10.1007/s11102-012-0397-5
32. Pivonello R, De Leo M, Cozzolino A, Colao A. The treatment of cushing’s disease. Endocr Rev (2015) 36(4):385–486. doi: 10.1210/er.2013-1048
33. Capatina C, Hinojosa-Amaya JM, Poiana C, Fleseriu M. Management of patients with persistent or recurrent cushing’s disease after initial pituitary surgery. Expert Rev Endocrinol Metab (2020) 15(5):321–39. doi: 10.1080/17446651.2020.1802243
34. Young J, Bertherat J, Vantyghem MC, Chabre O, Senoussi S, Chadarevian R, et al. Hepatic safety of ketoconazole in cushing’s syndrome: results of a compassionate use programme in France. Eur J Endocrinol (2018) 178(5):447–58. doi: 10.1530/EJE-17-0886
35. Pivonello R, Ferrigno R, De Martino MC, Simeoli C, Di Paola N, Pivonello C, et al. Medical treatment of cushing’s disease: an overview of the current and recent clinical trials. Front Endocrinol (Lausanne) (2020) 11(December). doi: 10.3389/fendo.2020.00648
Keywords: ketoconazole, Cushing’s disease, treatment, systematic review, meta-analysis
Citation: Viecceli C, Mattos ACV, Hirakata VN, Garcia SP, Rodrigues TdC and Czepielewski MA (2023) Ketoconazole as second-line treatment for Cushing’s disease after transsphenoidal surgery: systematic review and meta-analysis. Front. Endocrinol. 14:1145775. doi: 10.3389/fendo.2023.1145775
Received: 16 January 2023; Accepted: 07 April 2023;
Published: 08 May 2023.Edited by:
Monica Livia Gheorghiu, Carol Davila University of Medicine and Pharmacy, RomaniaReviewed by:
Leandro Kasuki, Instituto Estadual do Cérebro Paulo Niemeyer (IECPN), Brazil
Przemyslaw Witek, Warsaw Medical University, PolandCopyright © 2023 Viecceli, Mattos, Hirakata, Garcia, Rodrigues and Czepielewski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mauro Antônio Czepielewski, maurocze@terra.com.br
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
From https://www.frontiersin.org/articles/10.3389/fendo.2023.1145775/full

")
")
-scan-of-the-femoral-neck-showing-osteopenia")
-scan-of-the-lumbar-spine-showing-osteoporosis")











-Brain")
Adults with Cushing’s Syndrome Report High Burden Of Illness, Despite Ongoing Treatment
in News Items and Research
Posted
Key takeaways:
Adults with endogenous Cushing’s syndrome reported that the condition moderately affects their quality of life and causes them to have symptoms about 16 days in a given month, according to findings published in Pituitary.
“Our study aimed to evaluate the ongoing burden of Cushing’s syndrome in order to identify areas of unmet need,” Eliza B. Geer, MD, medical director of the Multidisciplinary Pituitary and Skull Base Tumor Center and associate attending of endocrinology and neurosurgery at Memorial Sloan Kettering Cancer Center, told Healio. “We found that patients with treated Cushing’s continue to experience ongoing symptoms more than half of the days in a given month, miss about 25 workdays per year and need twice the average number of outpatient visits per year, indicating a significant impact on daily function and work productivity. Some of these symptoms, like fatigue and pain, have not been well studied in Cushing’s patients, and need more attention.”
Geer and colleagues administered a cross-sectional survey to 55 adults aged 21 years and older who had been diagnosed with Cushing’s syndrome at least 6 months before the survey and were receiving at least one pharmacologic therapy for their disease (85% women; mean age, 43.4 years). The survey was conducted online from June to August 2021. Five patient-reported outcome scales were included. The CushingQoL was used to analyze quality of life, a visual analog scale was included to assess pain, the Brief Fatigue Inventory was used to measure fatigue, the Sleep Disturbance v1.0 scale assessed perceptions of sleep and the PROMIS Short Form Anxiety v1.0-8a scale was used to measure fear, anxious misery, hyperarousal and somatic symptoms related to arousal. Participants self-reported the impact of Cushing’s syndrome on daily life and their physician’s level of awareness of Cushing’s syndrome.
Some symptoms decline in severity over time
Of the study group, 81% had pituitary or adrenal tumors, and 20% had ectopic adrenocorticotropic hormone-producing tumors; 80% of participants underwent surgery to treat their Cushing’s syndrome.
The frequency of reported symptoms did not change from Cushing’s syndrome diagnosis to the time of the survey. The most frequently reported symptoms were weight gain, muscle fatigue and weakness and anxiety.
Participants reported a decline in symptom severity for weight gain, muscle fatigue and weakness and menstrual changes from diagnosis to the survey. Though symptom severity declined, none of the three symptoms were entirely eliminated. Adults did not report declines in severity for other symptoms. Hirsutism and anxiety were reported by few participants, but were consistently scored high in severity among those who reported it. There were no changes in patient satisfaction with medications from their first appointment to the time of the survey.
“It was surprising that anxiety and pain did not improve with treatment,” Geer said. “A quarter of patients at baseline reported anxiety and this percentage was exactly the same after treatment. Same for pain — nearly a quarter of patients reported pain despite treatment. While the presence of anxiety has been well-documented in Cushing’s patients, pain has not, and needs further study.”
Nearly half of primary care providers unable to diagnose Cushing’s syndrome
All participants reported having at least one challenge with being diagnosed with Cushing’s syndrome. Of the respondents, 49% said their primary care provider was unable to diagnose their Cushing’s syndrome and 33% initially received the wrong diagnosis. Physicians referred 49% of participants to a specialist, and 39% of adults said their doctor lacked knowledge or understanding of their condition.
The study group had a moderate level of quality of life impairment as assessed through the CushingQoL scale. The mean pain score was 3.6 of a possible 10, indicating low levels of pain. Moderate to severe levels of fatigue were reported by 69% of participants. Self-reported sleep and anxiety scores were similar to what is observed in the general population.
Participants said sexual activity, self-confidence and life satisfaction were most impacted by a Cushing’s syndrome diagnosis. Adults experienced symptoms a mean 16 days in a typical month and saw their outpatient physician an average of six times per year. Those who were employed said they miss 2 days of work per month, or about 25 days per year, due to Cushing’s syndrome.
“Longitudinal assessment of clinically relevant patient-reported outcomes based on validated measures and coupled with biochemical and treatment data is needed in a large cohort of Cushing’s patients,” Geer said. “This will allow us to identify clinically meaningful changes in symptom burden within each patient, as well as predictors of outcomes — which patients improve on which symptoms, and which patients do not feel better despite biochemical normalization. We need to improve our ability to help our patients feel better, not just achieve normal cortisol levels.”
For more information:
Eliza B. Geer, MD, can be reached at geere@mskcc.org.
From https://www.healio.com/news/endocrinology/20230830/adults-with-cushings-syndrome-report-high-burden-of-illness-despite-ongoing-treatment