

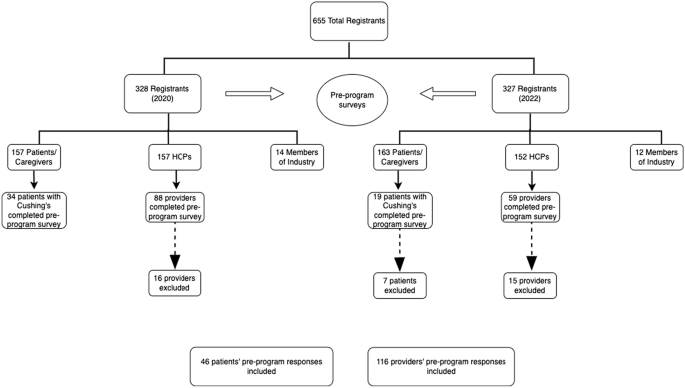


















Pituitary adenomas are adenohypophyseal tumors that can cause endocrinopathies, such as pituitary hormone hypersecretion or anterior hypopituitarism. Cell lineages are used to classify these tumors on the basis of immunohistochemical (IHC) staining of transcription factors, hormones, and other biomarkers.1 Pituitary adenomas differentiate from pluripotent stem cells along one of three lineage pathways, depending on the following active transcription factors (TFs): pituitary transcription factor 1 (PIT-1), T-box transcription factor (TPIT), or steroidogenic factor-1 (SF-1). Rarely, two or more discrete pituitary adenomas from different lineages are identified in patients; however, the etiology remains unclear.2 The incidence of multiple pituitary adenomas has been reported to be 1%–2% of all resected pituitary adenomas but is likely underestimated based on data from large autopsy series.1–4 Pluri-hormonal adenomas are also rare entities in which a single tumor contains multiple TF lineages with one or more hormonal excesses.1–3 Preoperative recognition of multiple or pluri-hormonal pituitary adenomas is rare, and most tumors are discovered incidentally upon autopsy, intraoperatively, or on histological analysis.2,3,5
In cases of multiple synchronous pituitary adenomas, only one hormone excess syndrome is most frequently evident on clinical presentation and endocrine workup. Silent pituitary tumors positive for prolactin on immunohistochemistry are the most prevalent additional, incidentally found tumor in cases of multiple pituitary adenomas.5 This is particularly true in Cushing disease.6,7 It is important to recognize the presence of multiple pituitary adenomas especially in the setting of hormonally active pituitary adenomas to provide optimal management for this subset of patients. Complete resection is curative for Cushing disease with the standard of care achieved through a transsphenoidal approach. Localization of the tumor presents a challenge because of suboptimal sensitivity of magnetic resonance imaging (MRI) in demonstrating microadenomas, the inconsistency of lateralization with inferior petrosal sinus sampling (IPSS), and delays in pathological analysis.1,8,9 Additionally, the presence of an additional pituitary adenoma can obscure the microtumor through its large size and mass effect and can act as a “decoy lesion” during MRI, IPSS, and resection.6
Consideration of multiple pituitary tumors is necessary for successful resection. In a patient with a biochemical picture of Cushing disease, the demonstration of an adenoma with negative adrenocorticotrophic hormone (ACTH) immunostaining and the absence of postoperative hypoadrenalism may indicate the existence of a double adenoma. Few cases have described a lack of remission of an endocrinopathy after transsphenoidal resection due to the presence of an additional adenoma,2,6,10 and even less so in the instance of the persistence of Cushing disease.6 We present a rare case of double pituitary adenomas in a patient presenting with Cushing disease who underwent two endoscopic endonasal transsphenoidal resections and immunostaining for prolactin and ACTH, respectively, with long-term normalization of her hypothalamic-pituitary-adrenal (HPA) axis.
Illustrative Case
History and Presentation
A 32-year-old female, gravida 0 para 0, with a history of a pituitary lesion and hyperprolactinemia presented to our institution for the evaluation for Cushing disease. Ten years earlier, the patient had presented to a gynecologist with hirsutism, galactorrhea, and oligomenorrhea. Her endocrine workup was remarkable for an elevated prolactin at 33.8 ng/mL (2.3–23.3 ng/mL), while follicle-stimulating hormone (FSH), luteinizing hormone (LH), and thyroid-stimulating hormone (TSH) levels were normal. No ACTH or cortisol levels were available. MRI demonstrated a 5 × 6 × 5–mm T1-weighted isointense pituitary lesion protruding into the suprasellar cistern due to a small sella size. She was treated with bromocriptine 2.5 mg daily for 5 years, with normalization of her prolactin level. Subsequent MRI demonstrated a stable lesion size and T1 and T2 hyperintensity in the region of the known pituitary lesion, considered to be posttreatment cystic change with proteinaceous contents and blood. After the normalization of her prolactin levels, she continued to have oligomenorrhea and abnormal hair growth. Polycystic ovaries were not visualized on ultrasound. She was started on oral contraceptives and then switched to the etonorgestrel implant.
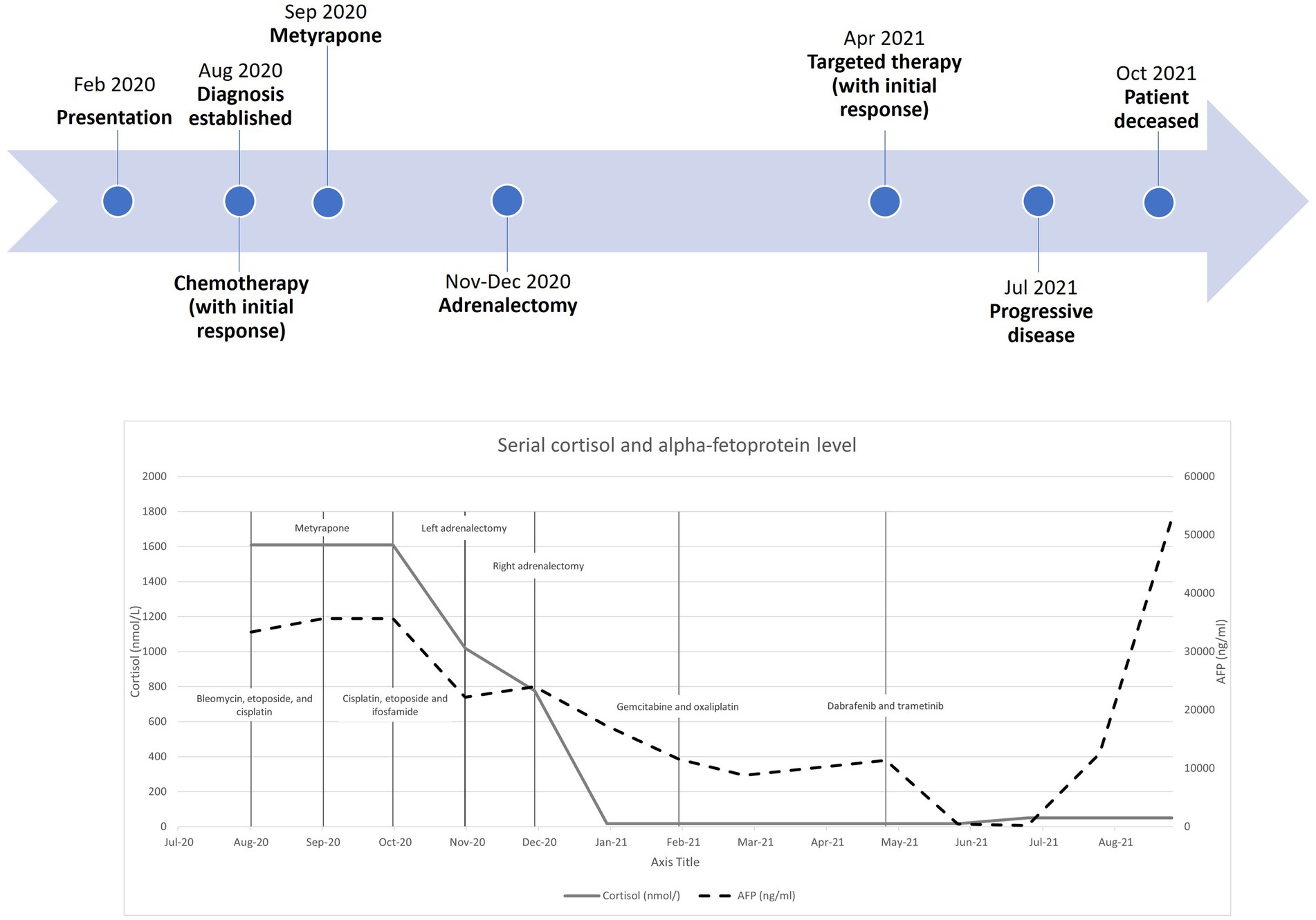
A decade after initial presentation, she presented to endocrinology at our institution with 3 years of weight gain, hirsutism, and potential oligomenorrhea. Vital signs were stable (blood pressure: 122/86; heart rate: 72 beats/min), and facial fullness and striae on her bilateral breasts were appreciated on physical examination. She was taking isoniazid and pyridoxine for a recent diagnosis of latent tuberculosis and had discontinued bromocriptine 5 years earlier. Her weight was 66.3 kg and body mass index (BMI) was 23.9 kg/m2. She reported that her maternal uncle had a pituitary tumor. Laboratory analysis was positive for elevated urinary free cortisol (UFC) of 109 µg per 24 hours (2.5–45 µg/24 h; Table 1) and nighttime salivary cortisol of 142 ng/mL (<100 ng/dL) with high-normal prolactin of 22.8 ng/mL (2.3–23.3 ng/dL) and normal FSH, LH, TSH, and thyroxine (T4). Dehydroepiandrosterone sulfate (DHEA-S) was 128 µg/dL (98.8–340.0 µg/dL). Imaging demonstrated a 4 × 4 × 4–mm pituitary lesion with decreased T1-weighted and increased central T2-weighted signal intensity in the left lateral pituitary (Fig. 1A–C). Desmopressin (Ferring Pharmaceuticals DDAVP) stimulation increased a basal ACTH of 49.9 pg/mL to ACTH of 91.2 pg/mL, and cortisol increased from 13.7 µg/dL to 21.2 µg/dL, consistent with neoplastic hypercortisolism. IPSS was performed, which showed a right-sided, central-to-peripheral ACTH gradient (Table 2). The patient elected to undergo endoscopic endonasal resection with the initial target as the left-lateral pituitary mass to achieve a cure for Cushing disease.
Urinary free cortisol at baseline and 3, 5, and 7 months after the primary resection
| Variable | Baseline | 3 Mos | 5 Mos | 7 Mos on Osilodrostat |
|---|---|---|---|---|
| Urinary free cortisol (4–50 µg/24 hrs) | 109 | 134.2 | 125.4 | 40.3 |
| Urinary creatinine (0.5–2.5 g/24 hrs) | 0.995 | 1.17 | 1.42 | 1.11 |
| Urinary vol (mL) | 1950 | 2300 | 2100 | 2125 |

Preoperative coronal precontrast (A) and postcontrast (B) T1-weighted magnetic resonance imaging (MRI) and T2-weighted MRI (C) demonstrated a 4-mm3 lesion (arrows) with decreased T1 and increased central T2 signal intensity in the left lateral pituitary. Two days after surgery, coronal precontrast (D) and postcontrast T1-weighted (E) and T2-weighted (F) MRI demonstrated the unchanged adenoma.
Preoperative inferior petrosal sinus sampling with corticorelin ovine triflutate 68 µg
| Time (mins) | ACTH (pg/mL) | Prolactin (ng/mL) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Peripheral | Petrosal Sinus | ACTH Ratio | Peripheral | Petrosal Sinus | Prolactin Ratio | |||||
| Rt | Lt | Rt | Lt | Rt | Lt | Rt | Lt | |||
| −5 | 50.6 | 225 | 1586 | 4.45 | 31.34 | 21 | 124 | 295 | 5.90 | 14.05 |
| 0 | 48.8 | 389 | 1376 | 7.97 | 28.20 | 22.2 | 185 | 198 | 8.33 | 8.92 |
| 3 | 69.8 | 4680 | 1333 | 67.05 | 19.1 | 22.1 | 396 | 32.5 | 17.92 | 1.47 |
| 5 | 80.9 | 4590 | 1623 | 56.74 | 20.06 | 22.1 | 436 | 32.2 | 19.73 | 1.46 |
| 10 | 112 | 4160 | 1660 | 37.14 | 14.82 | 20.2 | 367 | 42 | 17.90 | 2.05 |
ACTH or prolactin ratio = inferior petrosal sinus ACTH or prolactin/peripheral blood ACTH or prolactin.
Primary Resection and Outcomes
During the primary resection, abnormal tissue was immediately visible after a linear incision along the bottom of the dura, with an excellent plane of dissection. The inferomedial adenoma was distinct from the known left lateral lesion, and the resection was considered complete by the primary neurosurgeon. Subsequently, the left-sided adenoma was not pursued because of the historical prolactinoma diagnosis and an assumption that the newly discovered adenoma was the cause of ACTH hypersecretion. However, pathology of the inferomedial tumor was strongly and diffusely positive for prolactin (Fig. 2B), synaptophysin, and cytokeratin, with an Mindbomb Homolog-1 (MIB-1) proliferative index of 2.4%. ACTH, growth hormone (GH), FSH, LH, and TSH immunostaining were negative. TF immunohistochemistry was not available. On postoperative day (POD) 1, pituitary MRI was performed and demonstrated the unchanged 4-mm3 T1-weighted hypointense lesion with small central T2-weighted hyperintensity in the left lateral gland (Fig. 1D–F). Cortisol levels ranged from 9.7 to 76.2 µg/dL (4.8–19.5 µg/dL), and ACTH was 19.5 pg/mL (7.2–63.3 pg/mL) on POD 1.

Histological examination of surgical specimens from the inferomedial (A–C) and left lateral (D–F) lesions. The initial resection (hematoxylin and eosin [H&E], A) was strongly and diffusely positive for prolactin (B) with normal reticulin levels (C) indicating a lactotrophic pituitary adenoma. The second operation (H&E, D) was diagnostic for a corticotropic pituitary adenoma with diffusely positive adrenocorticotrophic hormone (ACTH) (E) and decreased reticulin (F). Original magnification ×100.
Early reoperation was discussed with the patient based on the pathology and persistent hypercortisolism; however, she elected to pursue conservative management with close follow-up. Postoperative cortisol nadir was 4.8 µg/dL (4.8–19.5 µg/dL) on POD 2 during her 4-day hospital stay. DHEA-S was significantly decreased from baseline at 22.3 µg/dL (98.8–340.0 µg/dL) and a prolactin level of 3.4 ng/mL (2.3–23.3 ng/dL) was low-normal. No glucocorticoids were administered during her hospital course. There was no clinical evidence of vasopressin deficiency while she was an inpatient.
Three months postoperatively, the patient reported insomnia, poor hair quality, fatigue, nocturnal sweating, and continued increasing weight gain with fat accumulation in the supraclavicular and dorsal cervical area. She had one spontaneous menstrual period despite the use of etonogestrel implant. UFC was increased at 134.2 µg/24 hours (4–50 µg/24 h; Table 1). The 8:00 am serum cortisol was 10.2 µg/dL (5.0–25.0 µg/dL). She was started on osilodrostat 2 mg twice daily for her persistent hypercortisolism, and she reported some clinical improvement; however, she had continued elevation in her late-night salivary cortisol levels up to 7.0 nmol/L. Other endocrine lab work was normal, with a prolactin of 13.5 ng/mL (2.8–23.3 ng/mL) and TSH of 3.67 µIU/mL (0.4–4.0 µIU/mL). Her weight had increased by 4.9 kg to 71.2 kg with a BMI of 25.3 kg/m2. Approximately 6 months postoperatively, she was amenable to a secondary resection targeting the remaining left lateral pituitary adenoma.
Secondary Resection and Outcomes
After obtaining adequate exposure for the secondary resection, the lesion in the left lateral aspect of the pituitary was targeted. The tumor was clearly identified and completely resected without intraoperative complication. IHC staining was diffusely positive for ACTH (Fig. 2E), synaptophysin, and cytokeratin with decreased reticulin and an MIB-1 index of 3.3%. Prolactin, GH, TSH, LH, and FSH immunostaining were negative. Postoperative cortisol monitoring demonstrated decreased levels, with a nadir of 2.0 µg/dL on POD 0. Levels of ACTH and DHEA-S were decreased at 4.4 pg/mL (7.2–63.3 pg/mL) and 13.3 µg/dL (98.8–340 µg/dL), respectively, on POD 1. Prolactin remained within the normal range at 8.2 ng/mL (2.8–23.3 ng/mL). The patient was started on intravenous hydrocortisone 50 mg every 8 hours for adrenal insufficiency. Postoperative symptoms of nausea, headache, and muscle weakness resolved with hydrocortisone administration. She was discharged on hydrocortisone 60 mg daily in divided doses for adrenal insufficiency and had no signs of vasopressin deficiency during her 2-day hospital course.
By 3 months, the patient reported decreased fatigue, myalgia, and insomnia and improved overall well-being and physical appearance. She was weaned down to a total daily dose of 20 mg of hydrocortisone and had lost 5.2 kg. Her menstruation returned while having an etonogestrel implant. Rapid ACTH stimulation was abnormal, with decreased cortisol at 30 minutes of 4.1 µg/dL (7.2–63.3 pg/mL) demonstrating continued adrenal insufficiency. Follow-up MRI demonstrated miniscule remaining left pituitary adenoma (Fig. 3). Seven months after her second surgery, she was started on 50 µg levothyroxine for primary hypothyroidism in the setting of slightly elevated TSH of 4.1 µIU/mL (0.4–4.0 µIU/mL) and a low-normal T4 of 0.8 ng/dL (0.7–1.5 ng/dL).

Postoperative imaging 3 months after the second operation demonstrates near gross-total resection (yellow arrows: surgical cavity) of the left lateral pituitary adenoma on coronal precontrast (A) and postcontrast T1-weighted (B) and T2-weighted (C) MRI.
Two years after the second resection, the patient lost 10.1 kg (weight, 61.1 kg; BMI, 21.76 kg/m2). Her ACTH stimulation test became normal, and hydrocortisone therapy was discontinued. At the 2-year time point, the patient and her husband successfully conceived a child.
Patient Informed Consent
The necessary patient informed consent was obtained in this study.
Discussion
Double or multiple pituitary adenomas are discovered in 0.37%–2.6% of resected pituitary lesions.3,4,6,11,12 A majority of multiple pituitary adenomas are not suspected before surgery with an inconclusive clinical presentation or endocrine laboratory workup.6 The presentation of multiple synchronous neoplasms is thought to be more common than having a single neoplasm with multiple lineages.1 Studies have shown that additional pituitary adenomas are seen at a rate of 1.6%–3.3% in Cushing disease in studies including both contiguous and noncontiguous double pituitary adenomas.6 Additional pituitary adenomas that are hormonally active make up 40% of resected double pituitary adenomas, with most staining for gonadotroph adenoma.13 Overall, the most common incidental pituitary adenoma is prolactinoma,6 which occurs most frequently with GH or ACTH adenomas.5 In very rare instances, Cushing cases can present with hyperprolactinemia and Cushing synchronously.6 Hormonal secretion and clinical presentation are variable, with the pathology most often attributed to only one component of double pituitary adenoma.3,14 The multiple-hit theory is the most common hypothesis for double pituitary adenoma etiology with coincidental monoclonal expansion of two or more lineages, which present with separate pseudo-capsules for each lesion.15
Observations
On presenting with Cushing disease, the differential diagnosis before the initial operation considered that the known left lateral pituitary adenoma could be a mixed tumor with both prolactin and ACTH lineages. Therefore, it was the initial target of the resection until discovering the second adenoma intraoperatively. With two distinct adenomas, the inferomedial adenoma was presumed to be the source of the ACTH hypersecretion and was subsequently resected. The left lesion was thought to be a prolactinoma and hormonally inactive after historical dopaminergic therapy and thus was not pursued during the initial surgery. However, pathology confirmed that the opposite was true. Few cases have also involved incidental pituitary tumors that look like the hormonally active adenoma and encourage resection of it, leaving the primary pituitary adenoma behind.6,7 It has been reported that these “decoy lesions” can cause surgical failure and require secondary operations.6,7,10,16 Intraoperative localization and confirmation of the adenoma classification may have also been helpful during the case, including tissue-based ACTH antibody assay,9 plasma ACTH measurements with a immunochemiluminometric method,17 or intraoperative ultrasound.5,6
The inferomedial second tumor was not appreciated or reported throughout her serial MRI studies from 2010 to 2020. Interestingly, imaging did demonstrate the left pituitary adenoma that was medically treated as a prolactinoma, although it was later diagnosed as an ACTH-secreting lesion on IHC staining. Preoperative visualization of a pituitary adenoma in Cushing disease is reported to be limited, with a reported 50% incidence with negative MRI with standard 1.5 T.1,18,19 MRI technical refinements in magnet strength, slice thickness, or enhanced spin sequences have increased sensitivity, but one-third of patients with Cushing disease still have negative scans.20 Small prolactinomas, especially those near the cavernous sinus, are also notoriously difficult to visualize on MRI, although recent advances using co-registration of 11C-methionine positron emission tomography–computed tomography with MRI (Met-PET/MRICR) may prove useful.21 Difficulty with preoperative visualization complicates a diagnosis of multiple adenomas, with or without multiple endocrinopathies, and negatively affects surgical planning. In a single-institution retrospective review of MRI in all cases of double pituitary tumors, only one of eight patients (12.5%) over 16 years of age had a positive MRI for double pituitary tumors and was diagnosed preoperatively.2
The patient’s preoperative IPSS demonstrated a right central-to-peripheral gradient. This was incongruent with the MRI demonstrating the single left-sided tumor. While IPSS is useful in confirming Cushing disease, its sensitivity for lateralization has been reported at only 59%–71%.9 With this in mind and a known left-sided adenoma on MRI, exploration of the right side of the pituitary was not originally planned. Ultimately, the left-sided adenoma was the source of ACTH hypersecretion, which remains incongruent with preoperative IPSS. It has been suggested that multiple pituitary adenomas in Cushing disease could further decrease its accuracy.1,6
The patient’s initial historical prolactin levels (33.8 ng/dL) were lower than reported levels of 100–250 ng/dL for microadenoma and >250 ng/dL in cases of macroadenoma. Normally, in active single prolactinoma, prolactin secretion is correlated to size. We do not suspect that the presence of more than one pituitary adenoma would affect the level of prolactin hypersecretion.6 Slight elevations in prolactin can be attributed to causes such as pituitary stalk effect, medications, and physiological stimulation. During the 5 years of bromocriptine therapy, the effect on the inferomedial prolactinoma was unknown, as it was not appreciated on MRI. There are reports of prolactinomas being less responsive to dopaminergic agonist therapy in cases of double adenomas.14,22 Upon resection of the inferomedial prolactinoma during the initial operation, there was no further change in the patient’s prolactin levels, which could most likely be attributed to prior dopaminergic therapy. Unfortunately, the initial endocrine laboratory workup did not include levels of ACTH or cortisol. In addition to hyperprolactinemia, Cushing disease can also present with changes in menstruation. After the secondary resection and removal of the ACTH-secreting pituitary adenoma, the patient’s oligomenorrhea resolved and she achieved pregnancy. Retrospectively, it remains unclear if the prolactinoma was once truly active hormonally.
Lessons
The rare presence of two pituitary adenomas can complicate the diagnosis, medical and surgical management, and long-term outcomes for patients. A complete endocrine workup is essential when a pituitary adenoma is suspected and can help screen for pluri-hormonal and multiple pituitary adenomas. In our patient, it is unknown when the onset of hypercortisolism was with the limited initial hormonal workup.
Currently, localizing and resecting the hormonally active adenoma in double or multiple pituitary adenomas remain a challenge, with limitations in preoperative imaging and intraoperative measures. After encountering the additional inferomedial lesion during surgery, resection of both adenomas during the initial surgery may have been prudent to ensure the resolution of Cushing disease. Although exploration for additional pituitary adenomas is not usually recommended, it could be considered in cases of multiple pituitary adenomas and uncertainty of the culprit of Cushing disease.
The current characterization of pituitary tumors by the World Health Organization includes immunohistochemistry for both transcription factors and pituitary hormones, with clinical usefulness to be determined by future studies. Multiple lineages can occur mixed in a single pituitary adenoma or across different noncontiguous adenomas and can only be determined by TF immunostaining. The left ACTH-staining lesion in our patient had some shrinkage and MRI changes, which may have been a response to dopaminergic therapy. Full characterization of the tumor cell lineages in this case remains undetermined without staining for TFs.
In conclusion, we report a rare case of Cushing disease concurrent with a prolactinoma leading to the need for repeat resection. This is one of the few reported cases of a double pituitary adenoma leading to a lack of biochemical remission of hypercortisolism after the initial surgery. Strategies for localization of the active tumor in double pituitary adenomas are essential for primary surgical success and the resolution of endocrinopathies.
Author Contributions
Conception and design: Zwagerman, Tavakoli, Shah, Findling. Acquisition of data: Zwagerman, Armstrong, Tavakoli, Shah, Ioachimescu, Findling. Analysis and interpretation of data: Zwagerman, Armstrong, Tavakoli, Shah, Coss, Ioachimescu, Findling. Drafting of the article: Zwagerman, Armstrong, Shah. Critically revising the article: Zwagerman, Armstrong, Tavakoli, Shah, Ioachimescu, Findling. Reviewed submitted version of the manuscript: Zwagerman, Armstrong, Tavakoli, Shah, Laing, Ioachimescu, Findling. Approved the final version of the manuscript on behalf of all authors: Zwagerman. Statistical analysis: Armstrong, Shah. Administrative/technical/material support: Zwagerman, Armstrong, Shah. Study supervision: Zwagerman, Tavakoli, Shah, Laing.
References
-
1↑
Asa SL, Mete O, Perry A, Osamura RY. Overview of the 2022 WHO Classification of Pituitary Tumors. Endocr Pathol. 2022;33(1😞6–26.
-
2↑
Roberts S, Borges MT, Lillehei KO, Kleinschmidt-DeMasters BK. Double separate versus contiguous pituitary adenomas: MRI features and endocrinological follow up. Pituitary. 2016;19(5😞472–481.
-
3↑
Mete O, Alshaikh OM, Cintosun A, Ezzat S, Asa SL. Synchronous multiple pituitary neuroendocrine tumors of different cell lineages. Endocr Pathol. 2018;29(4😞332–338.
-
4↑
Kontogeorgos G, Kovacs K, Horvath E, Scheithauer BW. Multiple adenomas of the human pituitary. A retrospective autopsy study with clinical implications. J Neurosurg. 1991;74(2😞243–247.
-
5↑
Budan RM, Georgescu CE. Multiple pituitary adenomas: a systematic review. Front Endocrinol (Lausanne). 2016;7:1.
-
6↑
Ratliff JK, Oldfield EH. Multiple pituitary adenomas in Cushing’s disease. J Neurosurg. 2000;93(5😞753–761.
-
7↑
Booth GL, Redelmeier DA, Grosman H, Kovacs K, Smyth HS, Ezzat S. Improved diagnostic accuracy of inferior petrosal sinus sampling over imaging for localizing pituitary pathology in patients with Cushing’s disease. J Clin Endocrinol Metab. 1998;83(7😞2291–2295.
-
8↑
Stroud A, Dhaliwal P, Alvarado R, et al. Outcomes of pituitary surgery for Cushing’s disease: a systematic review and meta-analysis. Pituitary. 2020;23(5😞595–609.
-
9↑
Erfe JM, Perry A, McClaskey J, et al. Long-term outcomes of tissue-based ACTH-antibody assay-guided transsphenoidal resection of pituitary adenomas in Cushing disease. J Neurosurg. 2018;129(3😞629–641.
-
10↑
Tolis G, Bertrand G, Carpenter S, McKenzie JM. Acromegaly and galactorrhea-amenorrhea with two pituitary adenomas secreting growth hormone or prolactin. A case report. Ann Intern Med. 1978;89(3😞345–348.
-
11↑
Magri F, Villa C, Locatelli D, et al. Prevalence of double pituitary adenomas in a surgical series: clinical, histological and genetic features. J Endocrinol Invest. 2010;33(5😞325–331.
-
12↑
Kim K, Yamada S, Usui M, Sano T. Preoperative identification of clearly separated double pituitary adenomas. Clin Endocrinol (Oxf). 2004;61(1😞26–30.
-
13↑
Mete O, Cintosun A, Pressman I, Asa SL. Epidemiology and biomarker profile of pituitary adenohypophysial tumors. Mod Pathol. 2018;31(6😞900–909.
-
14↑
Asa SL, Mete O, Riddle ND, Perry A. Multilineage pituitary neuroendocrine tumors (PitNETs) expressing PIT1 and SF1. Endocr Pathol. 2023;34(3😞273–278.
-
15↑
Zieliński G, Sajjad EA, Maksymowicz M, Pękul M, Koziarski A. Double pituitary adenomas in a large surgical series. Pituitary. 2019;22(6😞620–632.
-
16↑
Gonzalez A, Saindane AM, Neill SG, Oyesiku NM, Ioachimescu AG. The intriguing case of a double pituitary adenoma. World Neurosurg. 2019;126:331–335.
-
17↑
Raff H, Shaker JL, Nelson DK, Findling JW. Rapid measurement of corticotropin (ACTH) with a modified immunochemiluminescent assay. Clin Chem. 1994;40(7 Pt 1😞1344.
-
18↑
Buchfelder M, Nistor R, Fahlbusch R, Huk WJ. The accuracy of CT and MR evaluation of the sella turcica for detection of adrenocorticotropic hormone-secreting adenomas in Cushing disease. AJNR Am J Neuroradiol. 1993;14(5😞1183–1190.
-
19↑
Escourolle H, Abecassis JP, Bertagna X, et al. Comparison of computerized tomography and magnetic resonance imaging for the examination of the pituitary gland in patients with Cushing’s disease. Clin Endocrinol (Oxf). 1993;39(3😞307–313.
-
20↑
Grober Y, Grober H, Wintermark M, Jane JA, Oldfield EH. Comparison of MRI techniques for detecting microadenomas in Cushing’s disease. J Neurosurg. 2018;128(4😞1051–1057.
-
21↑
Bakker LEH, Verstegen MJT, Ghariq E, et al. Implementation of functional imaging using 11C-methionine PET-CT co-registered with MRI for advanced surgical planning and decision making in prolactinoma surgery. Pituitary. 2022;25(4😞587–601.
-
22↑
Coiré CI, Smyth HS, Rosso D, Horvath E, Kovacs K. A double pituitary adenoma presenting as a prolactin-secreting tumor with partial response to medical therapy. Case report. Endocr Pathol. 2010;21(2😞135–138.
From https://thejns.org/caselessons/view/journals/j-neurosurg-case-lessons/6/22/article-CASE23485.xml



![Three informative functional brain networks identified by the multivariate pattern classification method and the classification performance. a Three highly selected functional brain networks, including CerebN, FPN, and DMN, for differentiating active CD patients from NCs. b The frequency of the functional brain networks selected in the nested LOOCV experiments. c The receiver operating characteristic (ROC) curve (area under the ROC curve [AUROC] = 0.81) of the classification model built upon the selected most discriminative FNs. d The histogram of the classification rates of the permutation tests and the real classification rate. In panel (a), brain regions with significant functional connectivity were obtained by applying voxel-wise one-sample t tests to the IC’s z scores for each of the FNs across all active CD patients and NCs (p < 0.05, FWE corrected for multiple comparisons, and cluster size >400 voxels). CerebN, cerebellar network; FPN, fronto-parietal network; DMN, default mode network; CD, Cushing’s disease; Pres, CD patients before treatment (i.e., active CD patients); NCs, normal controls; FNs, functional networks; ICs, independent components; FWE, family-wise error; L, left; R, right.](https://karger.silverchair-cdn.com/karger/content_public/journal/nen/pap/10.1159_000534789/1/m_000534789_f02.jpeg?Expires=1704908362&Signature=vdfkKnXaQtQTdvWzKDqdSdEe2vovKN92cYHgj~2dIBm7zbIIjNsTfgHdYokETxkXD8GGkc9Xxl1FibTC1OvnjyDViPqo9gu7bANoUSB-y3gYfnfT4pZ7JCvBI2KUmro3eFAWGoukrGLDeiLFkV0~BCddOgCmbNQ4aLkCE9ZVdUkFxd941qGWGTRY7gFy-CE5BGCx68LObAMFdm0MIjKL6FOHwg6Up38MxjvIyZFZZ50i90ZKjMhL-R4J9MwHjgqyU22iU5yo9dZ9rOztGJlMeEToNiIPWzGkj-C5G121lO3HG81wTUaCE4YN4rdITBJwNwcJIjwVQYBcHwkLyhLKAw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)




-in-the-left-lower-lobe-with-ipsilateral-hilar-lymphadenopathy-and-pleural-effusion.")
.")

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Ectopic Adrenocorticotropic Hormone-Secreting Pheochromocytoma with Severe Metabolic Disturbances: A Case Report
in News Items and Research
Posted
Highlights
Abstract
Introduction
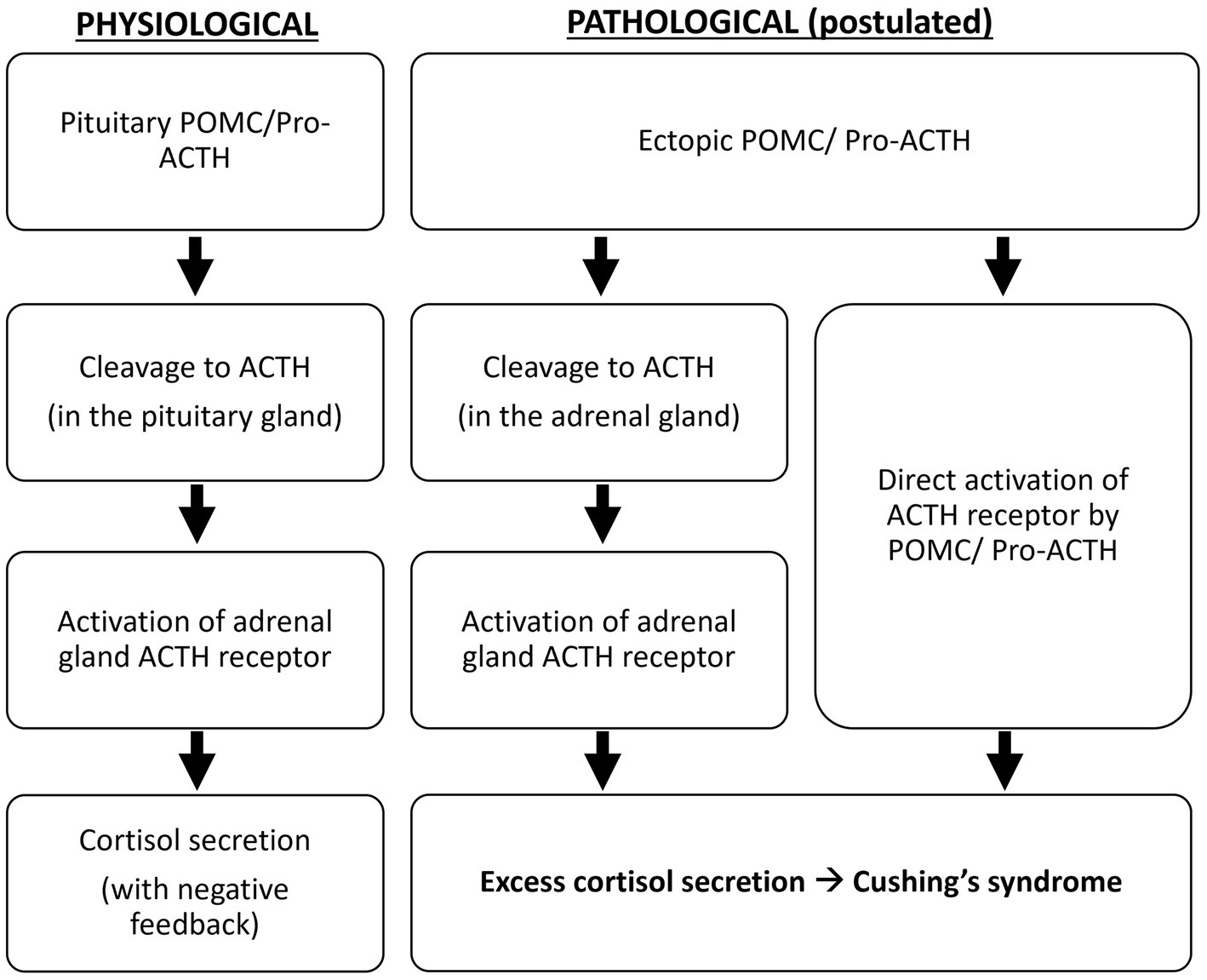
The occurrence of hypercortisolism resulting from adrenocorticotropic hormone (ACTH)-secreting pheochromocytoma is exceedingly uncommon, with limited documented instances thus far.
Presentation of case
We present a case of ectopic ACTH-secreting pheochromocytoma in a patient who suffered from severe metabolic disorders. Our clinical case outlines the diagnostic history, preoperative correction of the patient's metabolic disturbances and surgical strategy for management of a rare ectopic ACTH producing pheochromocytoma.
Discussion
Ectopic adrenocorticotropic hormone-secreting pheochromocytoma displays multifaceted clinical features and requires prompt diagnosis and multidisciplinary management in order to overcome the related severe clinical derangements.
Conclusion
The combination of biochemical and hormonal testing and imaging procedures is mandatory for the diagnosis of ectopic ACTH secretion, and in the presence of an adrenal mass, the possibility of an ACTH-secreting pheochromocytoma should be taken into account.
Keywords
1. Introduction
Neuroendocrine tumors such as Pheochromocytoma and paraganglioma (PPGL) are an uncommon occurrence. The prevalence of PPGL has been estimated to be between (2–8)/1 million, with a population rate of 1:2500–1:6500 [1], and it is associated with symptoms such as headache, irregular heartbeats, profuse sweating, high blood pressure, nausea, vomiting, nervousness, irritability, and a sense of imminent mortality [2]. Hypercortisolism is also a rare disorder with an incidence of 5/1 million, <10 % of patients with hypercortisolism are caused by ectopic secretion of ACTH [3], and these are most commonly seen in APUD tumors such as small cell bronchopulmonary carcinoma, pancreatic islet carcinoma, medullary thyroid carcinoma, pheochromocytoma, and melanoma [4]. Tumors that secrete both ACTH and catecholamines are much rarer. Here, we present a case of ectopic ACTH-secreting pheochromocytoma with severe metabolic disorders. The case report is compliant with SCARE Guidelines [5].
2. Case report
The patient is a 46-year-old male who presented to our hospital with recurrent symptoms of pheochromocytoma. He reported that he experienced unexplained symptoms such as panic attacks, headache, sweating, nausea, vomiting, and a feeling of imminent death, which could be alleviated by rest. His blood pressure was around 160–220/110–120 mmHg, and he was taking oral antihypertensive drugs regularly, with poor control of his blood pressure. The patient was admitted with a body temperature of 36.7 °C, heart rate of 130 beats/min, respiratory rate of 20 cycles per minute, blood pressure of 138/88 mmHg, height of 175 cm, weight of 67 kg, Body Mass Index (BMI): 21.88, normal physical examination, emaciated body type, thin subcutaneous fat, self-reported weight loss of 20 kg within 10 months, and history of diabetes mellitus of >1 year.
Laboratory tests showed that the blood potassium levels were within the normal range, while the blood sugar and beta-hydroxybutyrate levels were elevated (Table 1). Hormonal analysis showed plasma levels of free catecholamine and its metabolites were much higher than normal, in addition to a severe excess of cortisol secretion with circadian rhythm disorders and elevated serum ACTH (Table 2). Small dose dexamethasone suppression test (1 mg) yielded cortisol levels of over 1750 nmol/L (negative: no decrease in blood cortisol), thus confirming the presence of ACTH-dependent hypercortisolism. The results of electrocardiogram, chest computerized tomography (CT), cardiac ultrasound and thyroid ultrasound showed no obvious abnormality. Enhanced CT of the adrenal glands (Fig. 1) revealed the presence of a right adrenal tumor measuring approximately 5.3 ∗ 4.7 cm. Despite undergoing cranial MRI, no pituitary lesion was detected, thereby ruling out the possibility of Cushing's disease. The patient was further considered for possible ectopic ACTH syndrome and suspected ectopic ACTH-secreting pheochromocytoma.
Table 1. Laboratory test results.
Table 2. The patient's adrenal hormone results
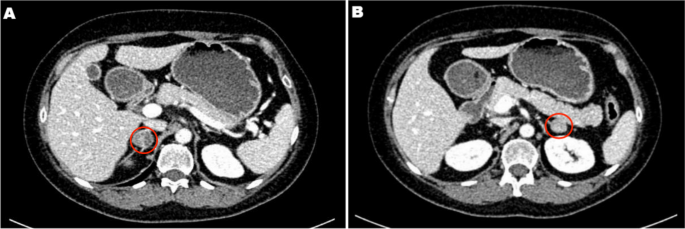
Fig. 1. Adrenal CT showed a 53 ∗ 47 mm mass in the right adrenal gland.
In response to the patient's pheochromocytoma symptoms and improve preoperative preparation, we used α-blocker (Phenoxybenzamine 20 mg q8h) to lower blood pressure and increase blood volume, antihypertensive medication (nifedipine 30 mg q12h, olmesartan tablets 20 mg q12h) to assist in lowering blood pressure, and β-blocker (metoprolol 47.5 mg q12h) to control the heart rate. On the 4th day in hospital, the patient was lethargic and had weak limbs. Urgent blood workup showed severe hypokalemia (2.85 mmol/L) as well as hyperglycemia (10.26 mmol/L). Patient was transferred to intensive care to correct intractable hypokalemia and diabetic ketoacidosis.
After the patient was transferred to ICU, a deep vein cannulation was performed with intravenous potassium chloride supplementation, and the patient's blood potassium was maintained at normal levels prior to surgery through a large amount of potassium supplementation (Fig. 2A). For diabetic ketoacidosis, insulin administration, rehydration, ketone elimination and other treatments were given and the amount of access was recorded, and it was found that the patient was polyuric, with the highest urine volume of 21,800 ml in a single day (Fig. 2B), and the amount of urine did not decrease by taking oral desmopressin tablets 0.1 mg bid.
Fig. 2. Changes in blood potassium and urine volume during the patient's hospitalization. A: Blood potassium level. B: Daily urine vlume.
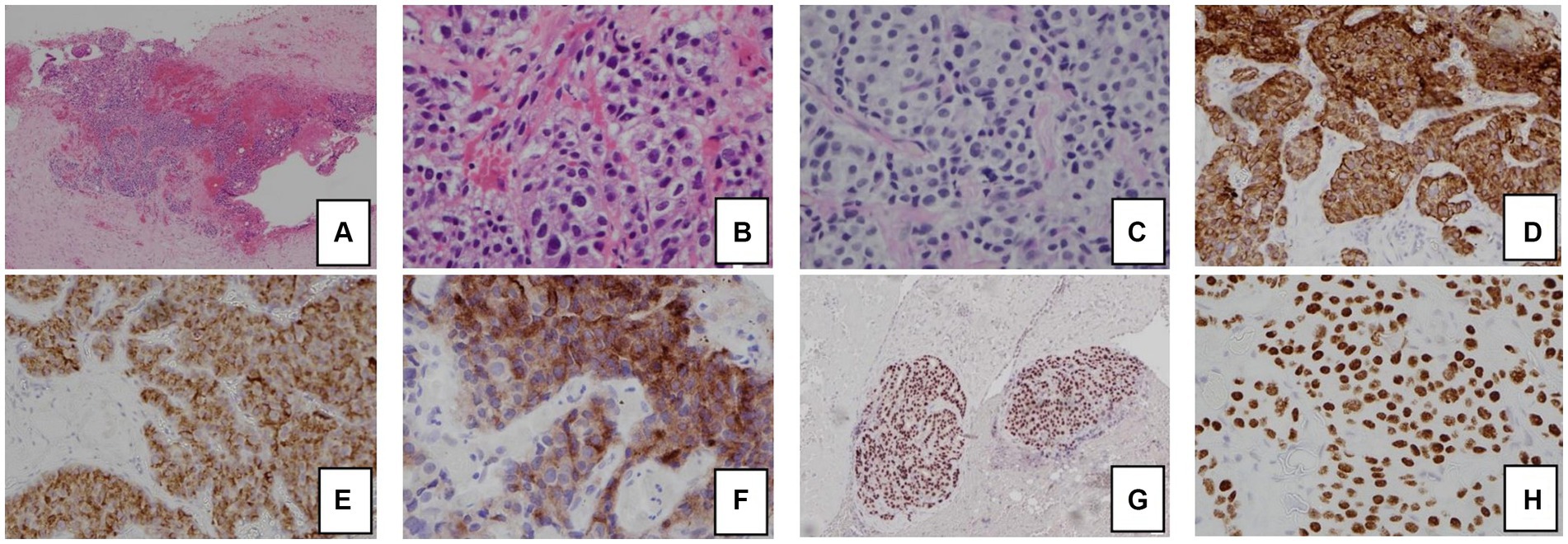
Eventually, the patient underwent laproscopic right adrenal tumor resection. Intraoperative changes in blood pressure and heart rate are shown in Fig. 3. On day 1 after surgery, the morning (8:00) ACTH level was 10.60 pg/ml, antihypertensive medications were discontinued, and his blood pressure was 100–120/60–90 mmHg. The patient's daily urine output and blood glucose gradually returned to normal levels after surgery. Pathology (Fig. 4😞 Adrenal pheochromocytoma with ACTH immunopositive staining, cellular heterogeneity was unremarkable, nuclear schizophrenic images were rare, no pericytes, choroidal invasion and necrosis were seen. The patient was discharged from the clinic in a satisfactory condition with adrenal insufficiency compensated by daily intake of Prednisone Acetate Tablets (20 mg), discontinued 6 months after surgery. No signs of recurrence were noted upon frequent follow-up examinations.
Fig. 3. Changes in patient's intraoperative blood pressure and heart rate.
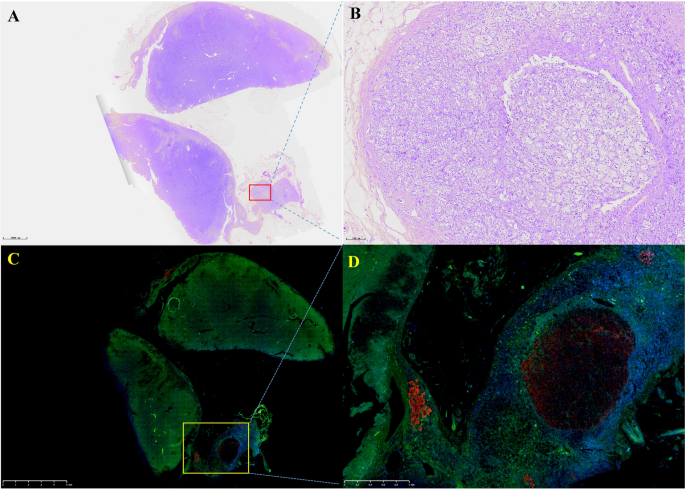
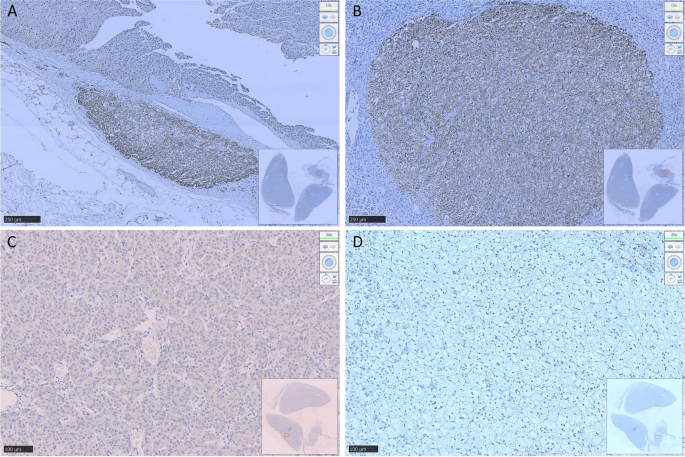
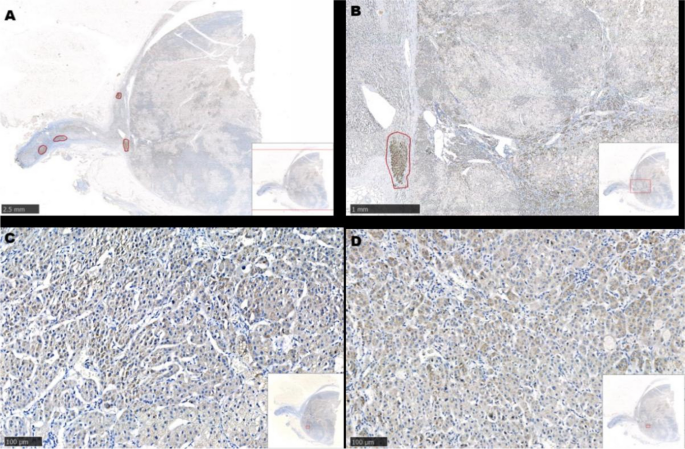
Fig. 4. Immunohistochemistry. A: hematoxylin and eosin staining B: ACTH.
3. Discussion
We share the management of a patient with ectopic ACTH-secreting pheochromocytoma with severe metabolic disturbances, where, in addition to the rare etiology, perioperative management of the clinical complications of catecholamines and hypercortisolism is very challenging [6].
Patients suffering from ectopic ACTH syndrome caused by pheochromocytoma commonly exhibit severe Cushing's syndrome (CS), significant diabetes mellitus, hypertension, and hypokalemia [7]. Additionally, a retrospective study revealed that the majority of patients presented with Cushing's syndrome [8], whereas another report indicated that only 30 % of patients presented with typical Cushing's syndrome, but weight loss was frequently observed [9]. Our patient's recent weight loss may be attributed to the body's hypermetabolic condition caused by catecholamines. Recent reports claim that catecholamines directly reduce subcutaneous and visceral fat [10]. Rapid onset of cortisolism appears to be a feature of ACTH-secreting pheochromocytomas, because of the rapid onset of severe hypercortisolism, and our patient did not exhibit typical Cushing's symptoms [8].
Despite the absence of typical Cushing-like symptoms, this patient displayed persistent hypokalemia, a prevalent metabolic manifestation of Cushing's syndrome, particularly in ectopic ACTH syndrome, where hypokalemia is observed in 74 %–95 % of patients, in contrast to 10 % of patients with Cushing's disease [11]. Glucocorticoids have the ability to interact with aldosterone receptors, resulting in specific aldosterone-like reactions, while ectopic ACTH syndrome typically generates a higher amount of cortisol compared to Cushing's disease, ultimately causing more pronounced hypokalemia [7]. The perioperative management of patients with ACTH-secreting pheochromocytomas poses a significant challenge due to severe hypokalemia, and our patient's potassium levels remained within the normal range through extensive central venous potassium supplementation, without the need for cortisol secretion inhibition medications.
The severity of hypertension in patients with ACTH-secreting pheochromocytomas seems to surpass that of patients with pheochromocytomas alone [12]. Hypercortisolism amplifies catecholamine-induced hypertension [13]. In the case of hypertension in patients with pheochromocytomas, alpha-blockers are favored for reducing blood pressure and enlarging blood volume, while for individuals whose blood pressure is not adequately managed with alpha-blockers alone, a combination of medications is recommended. Proper preoperative readiness for expanding the volume is crucial for a successful surgical procedure. Patients with ACTH-secreting pheochromocytoma have a greater prevalence and intensity of diabetes mellitus compared to those with pheochromocytoma alone [14], and our patient displayed a combination of severe diabetes mellitus and ketoacidosis. Insulin exhibits swift action and adaptable dosage, effectively averting hypoglycemia and effectively addressing hyperglycemia, rendering it the preferred medication for regulating blood glucose levels in individuals with ectopic CS [6].
Managing the water-electrolyte balance in this patient proved to be an arduous task, and the diabetes insipidus may have been one of the complications, with a maximum urine output of 21,800 ml in a single day (Fig. 2), and we hold the belief that the patient's diabetes insipidus is caused by a range of factors, such as hypokalemia, hypercortisolism, and severe diabetes mellitus. Indeed, hypokalemia may cause renal impairment, which reduces the ability to concentrate urine and lack of response to antidiuretic hormone (ADH), leading to nephrogenic diabetes insipidus [15]. Cortisol increases renal plasma flow and glomerular filtration rate, and also inhibits the secretion of antidiuretic hormone, leading to neurogenic diabetes insipidus [16].
For hypercortisolism, surgery to target the cause is the first-line treatment, and surgical removal of primary tumor may lead to 40 % radical treatment and 80 % complete remission of ectopic ACTH syndrome [17].
4. Conclusion
Preoperative diagnosis and management of pheochromocytoma, an extremely rare cause of ectopic ACTH syndrome, is challenging. Proper preoperative recognition of complications of both hypercortisolism and catecholamines excess is the key to prevent the morbidity and mortality of an ACTH-producing pheochromocytoma. If diagnosed successfully and managed intensively, they are curable.
Consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Ethical approval
Shandong Provincial Hospital Affiliated to Shandong First Medical University does not require ethical approval for publication of case reports. Signed consent from the patient has been received.
Funding
No funding was received for this research.
Author contribution
Shangjian Li: study concept or design, data collection, data analysis or interpretation, writing the paper
Xudong Guo: study concept or design, data collection, data analysis or interpretation, writing the paper
Hanbo Wang: study concept or design, data analysis or interpretation
Ni Suo: study concept or design, data analysis or interpretation
Xiuqing Mi: study concept,data collection
Shaobo Jiang: study concept or design, data analysis or interpretation, writing the paper
Guarantor
Shangjian Li
Xudong Guo
Shaobo Jiang
Conflict of interest statement
All authors declare no conflict of interest.
Acknowledgements
None.
References
This article is free to access.
This article is free to access.