

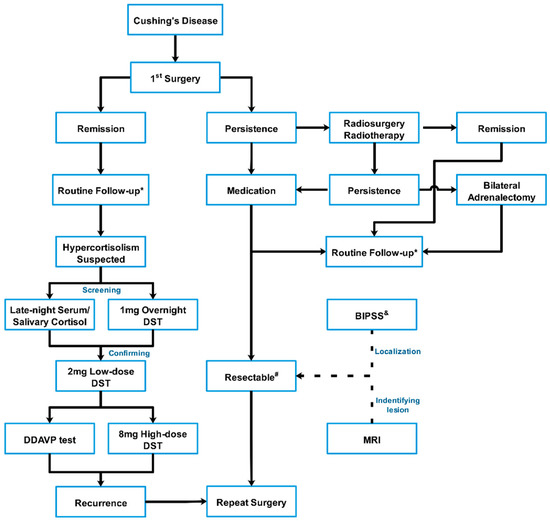
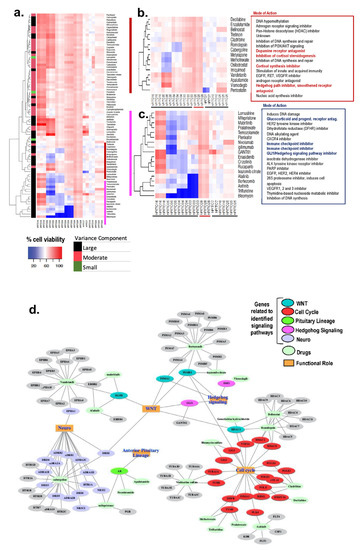
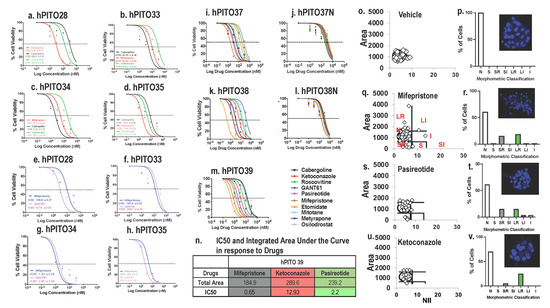
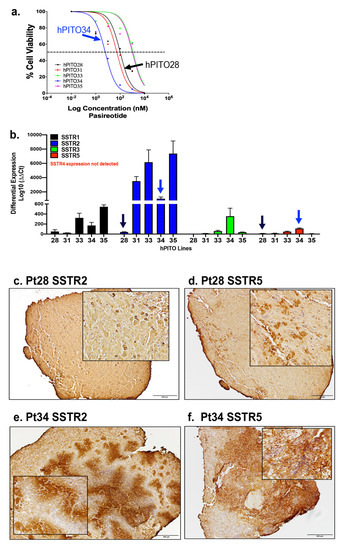
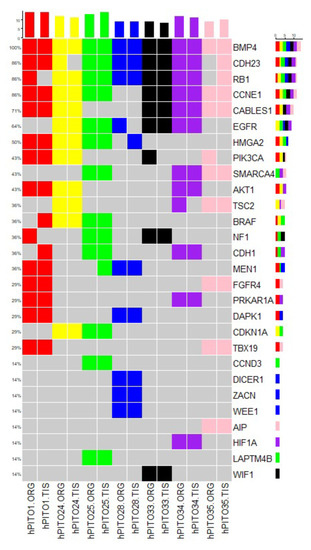
![Enrollment and follow-up of the study population after the adrenalectomy. aThe prevalence of patients with overt hypercortisolism and hypertension among this study population may be higher than in the general population and therefore needs to be carefully interpreted given that the study institute is one of the largest centers for adrenal diseases in Japan. bAll patients in this category showed autonomous cortisol secretion (ie, serum cortisol levels >5.0 µg/dL [138 nmol/L] after a 1-mg dexamethasone suppression test). cOne case underwent adrenalectomy at another hospital and therefore no information was available after the adrenalectomy. dThe adrenalectomy was performed for 33 patients who were expected to improve their clinical symptoms and/or metabolic disorders, including hypertension. This assessment was mainly based on autonomous cortisol secretion evaluated by a 1-mg dexamethasone suppression test, complicated metabolic disorders, and autonomous aldosterone secretion evaluated by adrenal venous sampling for patients who were positive for the screening and confirmatory tests of primary aldosteronism. Details in the assessment can be found in the Methods section or elsewhere [18-20].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jes/7/1/10.1210_jendso_bvac167/2/m_bvac167f1.jpeg?Expires=1673226229&Signature=lH6Fo2svnZ4zLJ1vgDEWEShEHMnJYbw7JNP1Va6L~hR1SEu38yoBriwSZN9YHqMv3KcleFNJHIJun5Q0YYsBF9KclE-FpKJ0hmyzTCE6ZPxtRFpYNYDFSPY~4HTccGaYS6NvslM7g4mleOc1ZalfFRPRAzNZAeIvsLeaKXeF4pXP5dqL0W1jT-SgLyVkPbVhteCGMSm4fLytXM85yaB0AhExA3WwQuihmQU1sYbI0BeOE5tneusxGAFbxZDx2CbCT1UuYvaYHNAYQ6Q7NHMF2cpE0fW9HSkUpejOb5zgycvPAFaPiWmDZxoHDDhCIVzuUFpr7ngYe7MoS2O-QKd6tA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)








As part of a small 2019 deal, Italian drugmaker Recordati snagged a trio of underperforming Novartis endocrinology meds, including a late-stage candidate for Cushing's disease. Less than a year later, that drug is cleared for market after an FDA green light.
The FDA based its review on phase 3 data showing 86% of patients treated with Isturisa showed normal cortisol levels in their urine after eight weeks, compared with 29% of patients treated with placebo, the drugmaker said.
Recordati is "actively building its commercial, medical, and market access teams" to accommodate Isturisa's launch through its recently created U.S. endocrinology business unit, it said. The drugmaker will launch the drug with a "comprehensive distribution model" through specialty pharmacies.
Novartis, once the owner of Isturisa, turned the asset over to Recordati in 2019 as part of a $390 million offload of some of the Swiss drugmaker's endocrinology portfolio.
Recordati received Signifor, long-acting sister Signifor LAR and Isturisa, positioned as a successor drug to Signifor. The purchase included milestone payments tied to Isturisa.
Recordati talked up the buy of the Cushing's disease trio as a boon for its rare disease portfolio, calling it a "key and historical milestone" at the time.
From https://www.fiercepharma.com/pharma/recordati-scores-fda-nod-for-cushing-s-disease-med-isturisa











{kind=link}
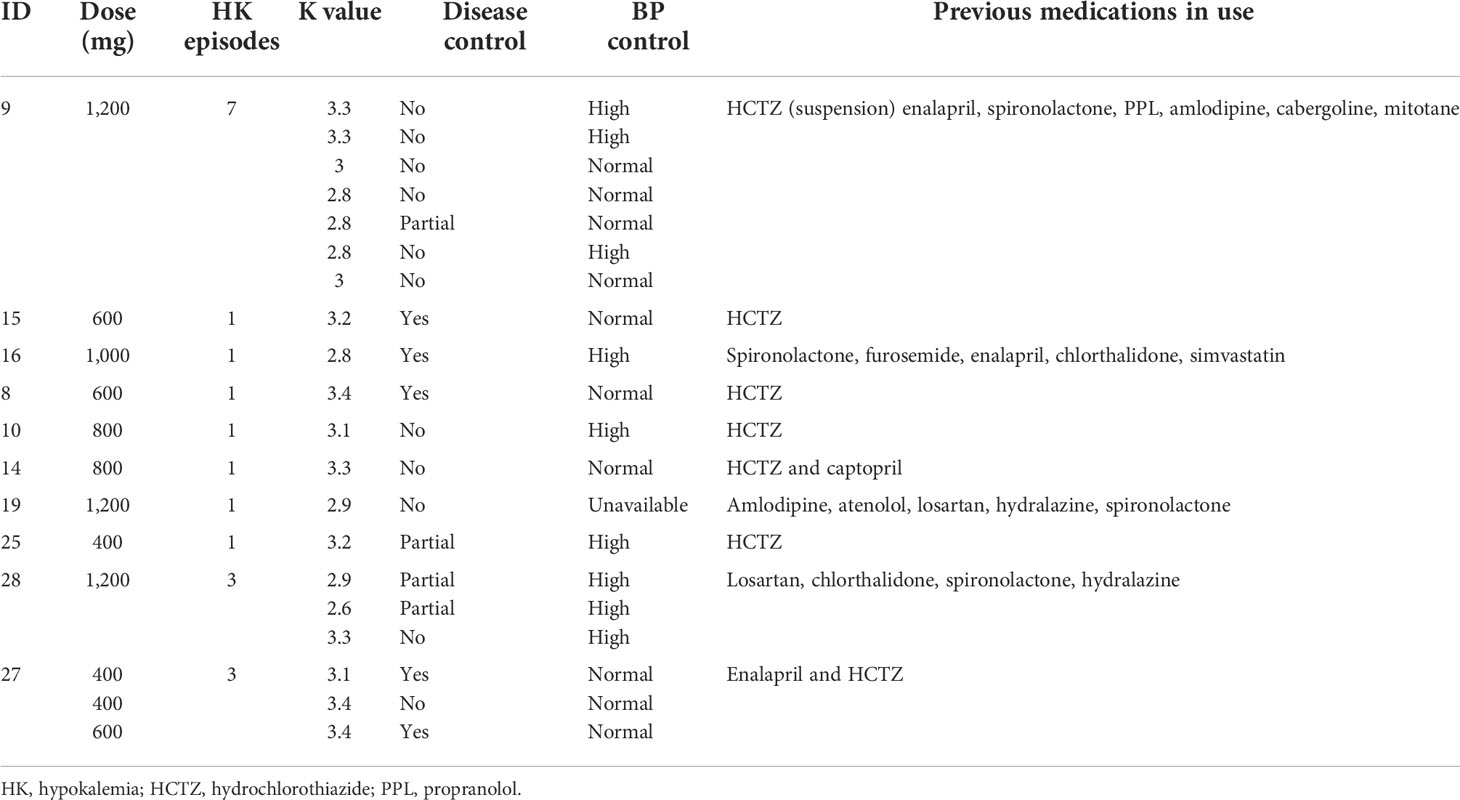
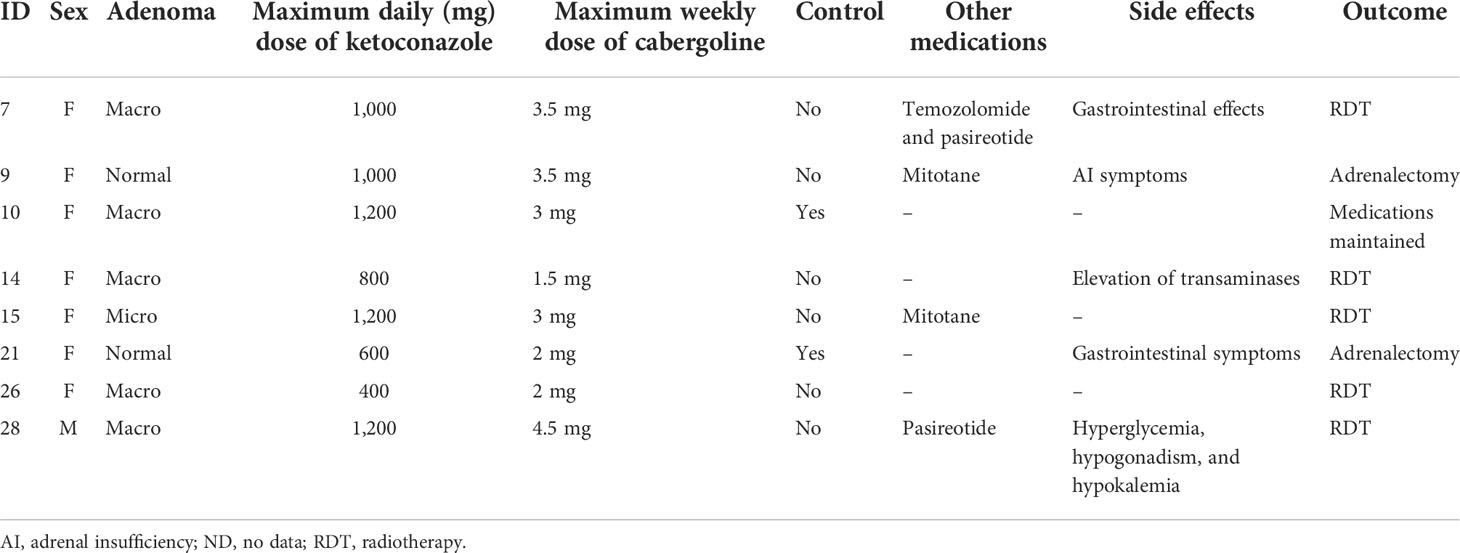
CRH Stimulation Test Boosts Cushing Disease Diagnosis
in News Items and Research
Posted
The study covered in this summary was published on Research Square as a preprint and has not yet been peer reviewed.
Key Takeaways
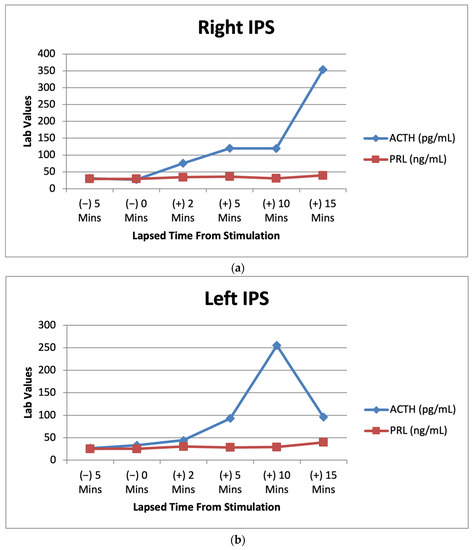
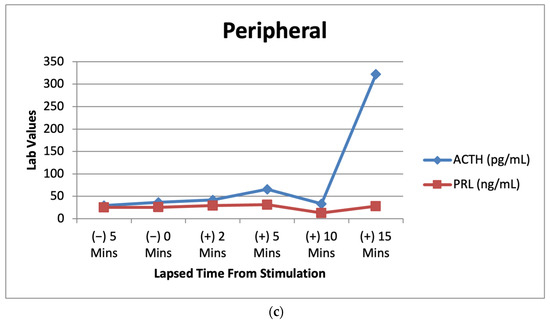
Adding a corticotropin-releasing hormone (CRH) stimulation test immediately following a 2-day low-dose dexamethasone suppression test (LDDST) ― what's known as a Dex-CRH test and was first introduced in 1993 ― identified Cushing disease in 5 of 65 people (7.7%) with a confirmed diagnosis but who had previously shown normal cortisol levels on a conventional LDDST.
However, the Dex-CRH test also resulted in one (2.5%) false positive case compared with an LDDST alone.
Measuring serum dexamethasone levels further improved the diagnostic accuracy of the Dex-CRH test.
Why This Matters
It can be challenging to diagnose Cushing syndrome and to differentiate Cushing disease from nonneoplastic physiologic hypercortisolism caused by conditions that can present with Cushing syndrome–like clinical features, such as diabetes and obesity.
The Dex-CRH test, first described in 1993, initially appeared superior to an LDDST alone for ruling out nonneoplastic hypercortisolism, with a report of 100% sensitivity, specificity, and diagnostic accuracy. However, subsequent studies that used different protocols and in which dexamethasone was not measured had results that called into question the accuracy, sensitivity, and specificity of the Dex-CRH test.
This study reports the accuracy, sensitivity, and specificity of the Dex-CRH test for diagnosing Cushing disease, performed as first described, in 107 patients, including 74 for whom dexamethasone was also measured.
Study Design
The researchers analyzed data from 107 patients with suspected Cushing disease who underwent a Dex-CRH test during 2002–2014 at the Cleveland Clinic.
Key Results
Sixty-five people received a confirmed diagnosis of Cushing disease and underwent follow-up for a median of 66 months. Cushing disease was not confirmed in 42 patients who were followed for a median of 52 months.
The median age of the 107 patients was 40 years, and 82% to 88% were women. The median body mass index for these patients was 34–37 kg/m2.
Among the 65 patients with confirmed Cushing disease, five patients (7.7%) had a suppressed cortisol level no greater than 1.4 μg/dL after the LDDST but were appropriately classified as having Cushing disease with a cortisol level that surpassed 1.4 μg/dL by 15 minutes after CRH stimulation.
In contrast, 3 of 42 patients (7.1%) in the group without confirmed Cushing disease had an abnormal Dex-CRH test result. For one of these three patients, the LDDST result was borderline normal, with a cortisol level post-DEX of 1.4 μg/dL that increased to 3.1 μg/dL by 15 minutes after CRH stimulation, which resulted in this patient receiving a false positive diagnosis.
A cortisol threshold value of more than 1.4 μg/dL during the Dex-CRH test was diagnostic of Cushing disease with sensitivity of 100%, specificity of 93%, and diagnostic accuracy of 97%.
Among the 74 patients with dexamethasone measurements, the sensitivity of the Dex-CRH test was unchanged, but the specificity and diagnostic accuracy increased to 97% and 99%, respectively.
Limitations
The study was retrospective.
Not all patients underwent measurement of dexamethasone level.
No uniform protocol existed for the diagnostic work-up and follow-up of patients suspected of having Cushing disease.
Disclosures
The study did not receive commercial funding.
The authors had no financial disclosures.
This is a summary of a preprint research study , "The Addition of Corticotropin-Releasing Hormone to 2-Day Low Dose Dexamethasone," written by researchers primarily from the Cleveland Clinic and Johns Hopkins University School of Medicine, published on Research Square, and provided to you by Medscape. This study has not yet been peer reviewed. The full text of the study can be found on research square.com.
From https://www.medscape.com/viewarticle/985984