










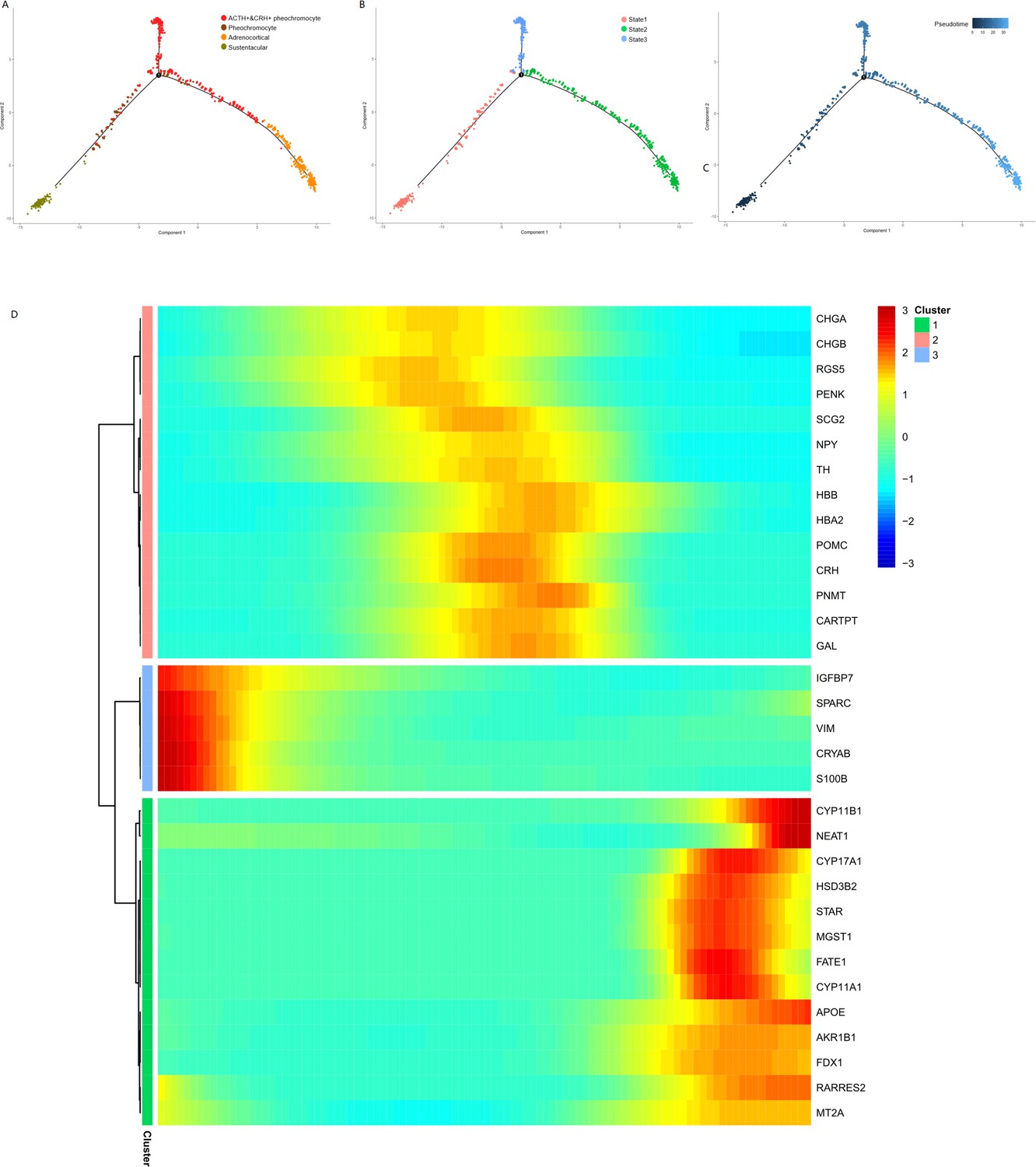

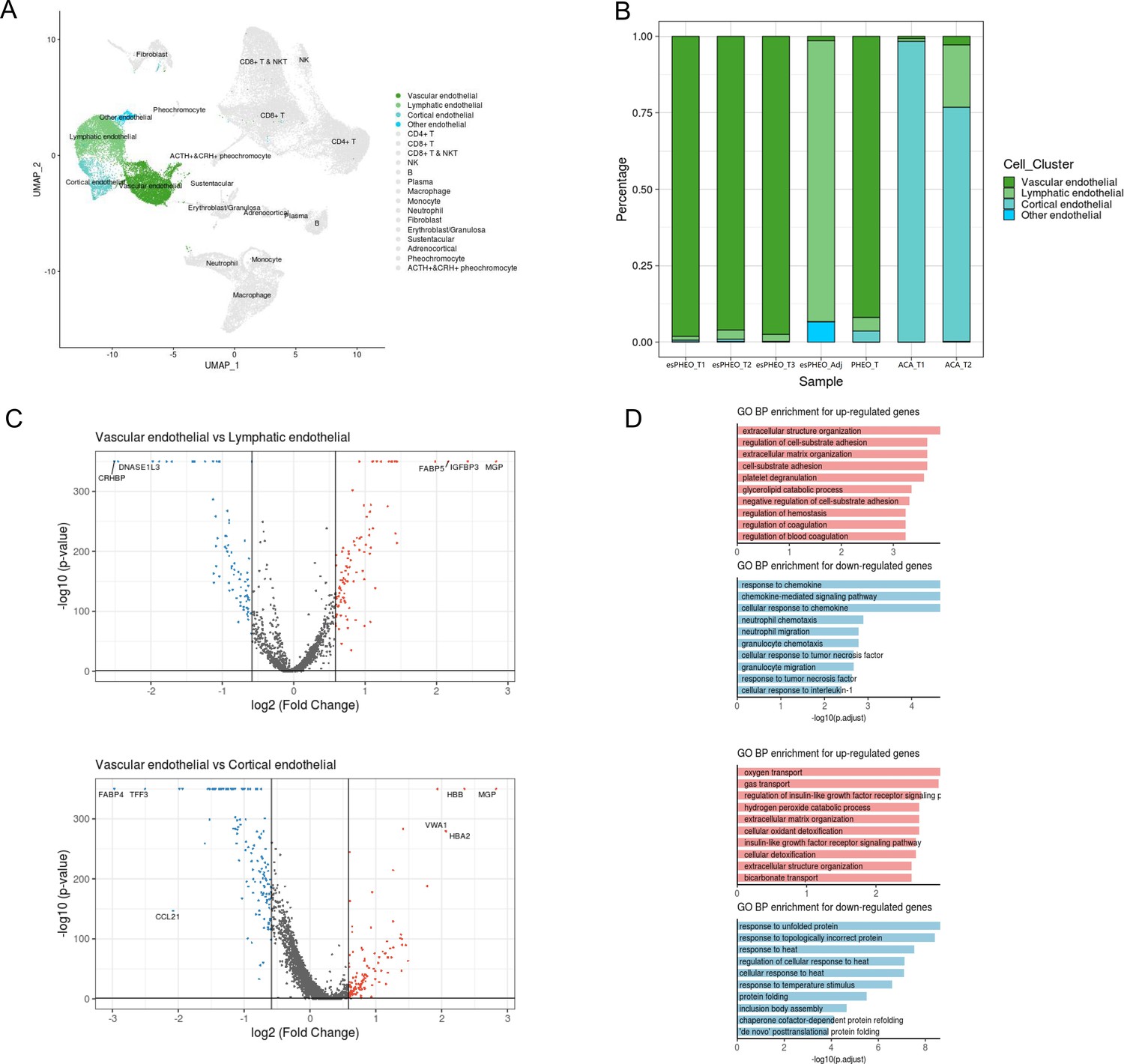
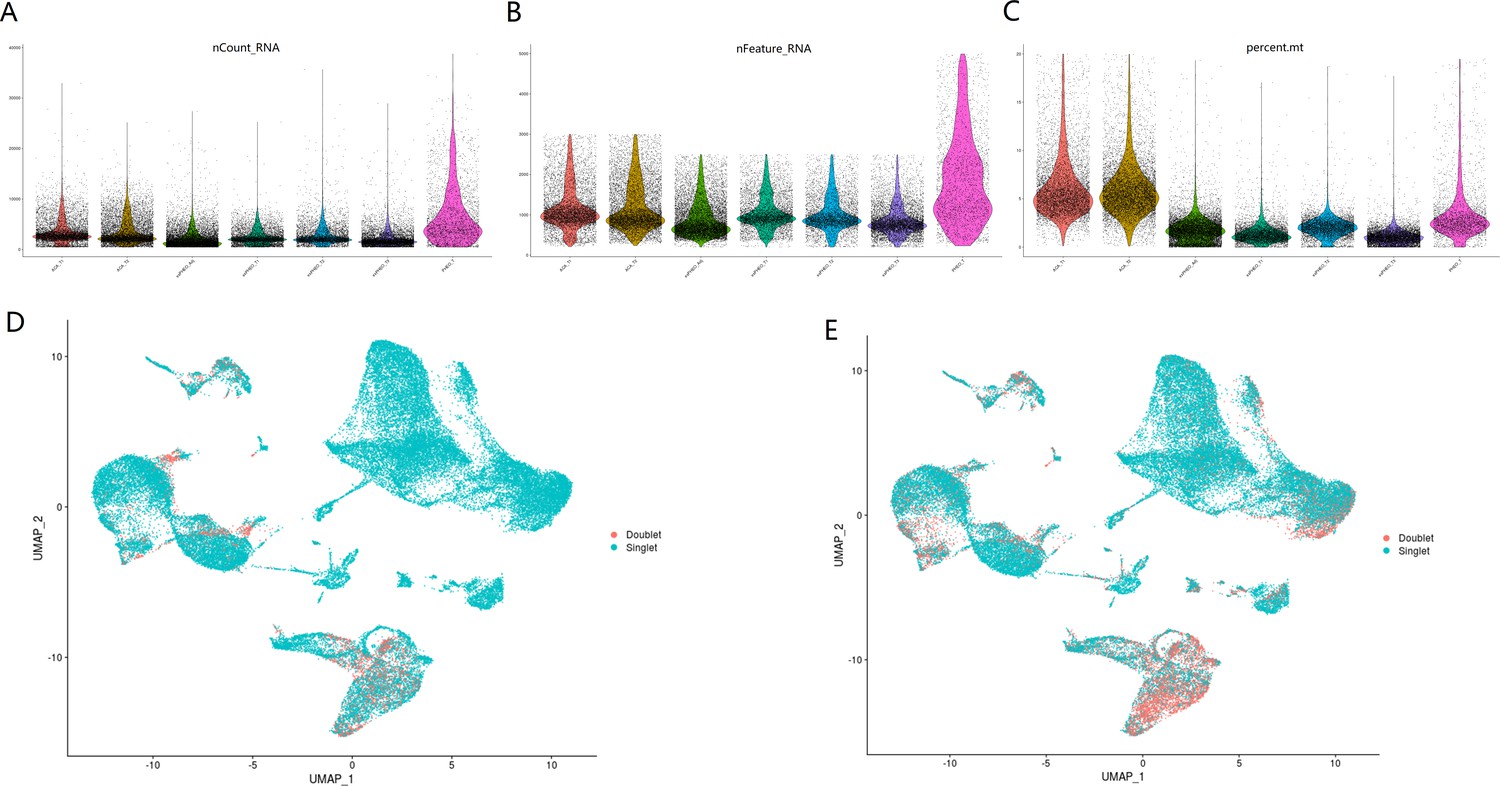
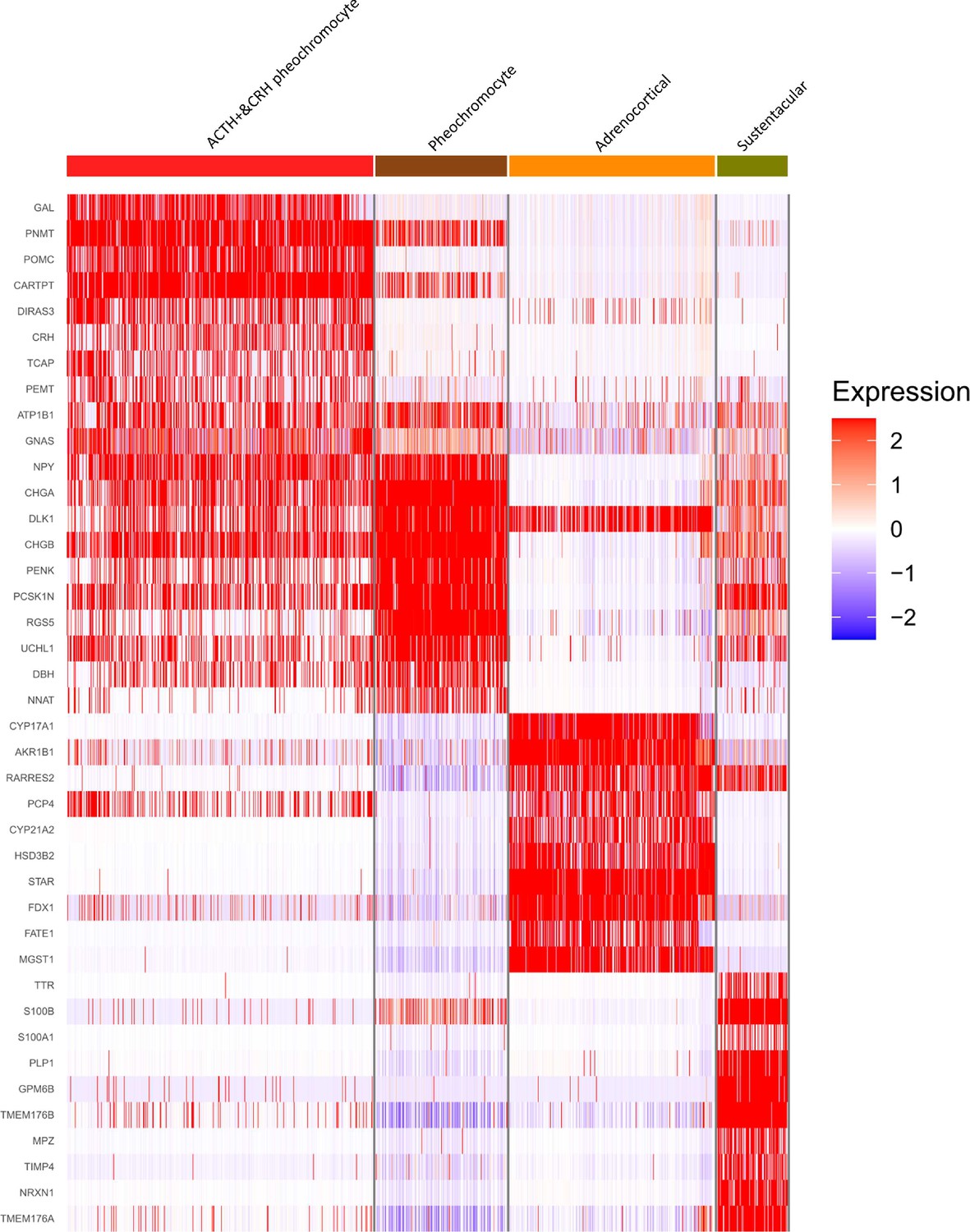


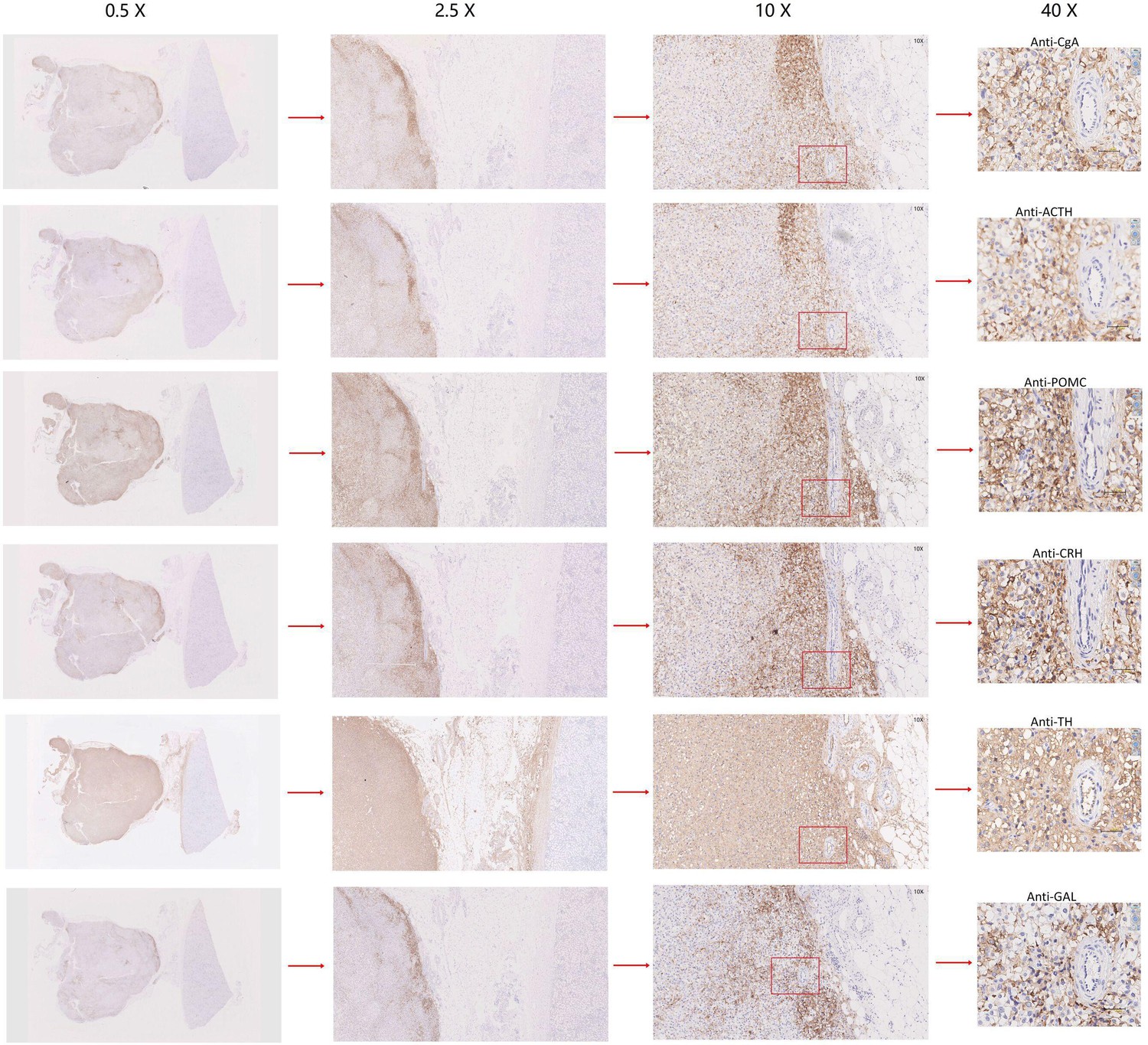

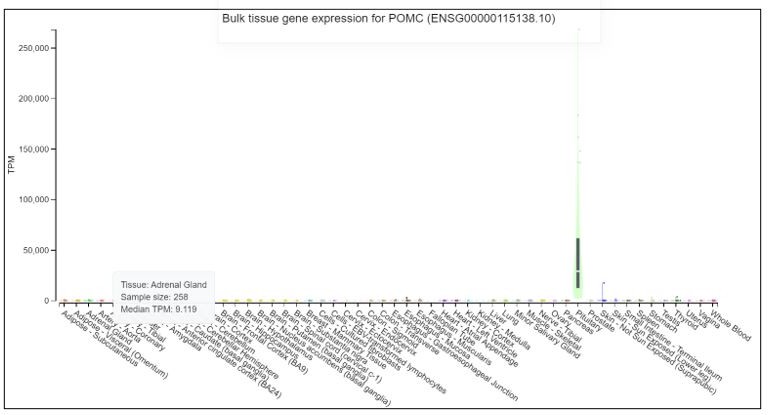













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Patient: Female, 74-year-old
Final Diagnosis: ACTH-dependent Cushing’s syndrome • ectopic ACTH syndrome
Symptoms: Edema • general fatigue • recurrent mechanical fall
Medication: —
Clinical Procedure: —
Specialty: Critical Care Medicine • Endocrinology and Metabolic • Family Medicine • General and Internal Medicine • Nephrology • Oncology
Objective:
Unusual clinical course
Background:
Adrenocorticotropic hormone (ACTH)-dependent Cushing’s syndrome (CS) secondary to an ectopic source is an uncommon condition, accounting for 4–5% of all cases of CS. Refractory hypokalemia can be the presenting feature in patients with ectopic ACTH syndrome (EAS), and is seen in up to 80% of cases. EAS can be rapidly progressive and life-threatening without timely diagnosis and intervention.
Case Report:
We present a case of a 74-year-old White woman who first presented with hypokalemia, refractory to treatment with potassium supplementation and spironolactone. She progressively developed generalized weakness, recurrent falls, bleeding peptic ulcer disease, worsening congestive heart failure, and osteoporotic fracture. A laboratory workup showed hypokalemia, hypernatremia, and primary metabolic alkalosis with respiratory acidosis. Hormonal evaluation showed elevated ACTH, DHEA-S, 24-h urinary free cortisol, and unsuppressed cortisol following an 8 mg dexamethasone suppression test, suggestive of ACTH-dependent CS. CT chest, abdomen, and pelvis, and FDG/PET CT scan showed a 1.4 cm right lung nodule and bilateral adrenal enlargement, confirming the diagnosis of EAS, with a 1.4-cm lung nodule being the likely source of ectopic ACTH secretion. Due to the patient’s advanced age, comorbid conditions, and inability to attend to further evaluation and treatment, her family decided to pursue palliative and hospice care.
Conclusions:
This case illustrates that EAS is a challenging condition and requires a multidisciplinary approach in diagnosis and management, which can be very difficult in resource-limited areas. In addition, a delay in diagnosis and management often results in rapid deterioration of clinical status.
Background
Cushing’s syndrome (CS) has a variety of clinical manifestations resulting from excess steroid hormone production from adrenal glands (endogenous) or administration of glucocorticoids (exogenous) [1,2]. Endogenous CS is classified into 2 main categories: ACTH-dependent and ACTH-independent disease. In ACTH-dependent disease, the source of ACTH can further be subdivided into either the pituitary gland or an ectopic source [2]. Ectopic ACTH syndrome (EAS) results from excess production of ACTH from extra-pituitary sources [2] and accounts for approximately 4–5% of cases of CS [3,4]. Common clinical manifestations of CS include weight gain, central obesity, fatigue, plethoric facies, purple striae, hirsutism, irregular menses, hypertension, diabetes/glucose intolerance, anxiety, muscle weakness, bruising, and osteoporosis [2]. Hypokalemia is a less defining feature, seen in roughly 20% of cases with CS. However, it is present in up to 90% of cases with EAS [2,5], which is attributed to the mineralocorticoid action of steroid [6].
Hypercortisolism due to EAS is usually severe and rapid in onset, and excess cortisol levels can lead to severe clinical manifestations, including life-threatening infections [7]. Moreover, in most patients with EAS, the source of excess ACTH is an underlying malignancy that can further result in rapid deterioration of the overall clinical condition. Although numerous malignancies have been associated with EAS, lung neuroendocrine tumors (NETs) are the most common [2,8]. Since the treatment of choice for EAS is complete resection of the tumor, the correct localization of the source of ectopic ACTH is crucial in managing these patients. Traditional radiological investigations can localize these tumors in up to 50% of cases [9]; however, recent studies utilizing somatostatin receptor (SSTR) analogs have increased the sensitivity and specificity of tumor localization [9–11]. This case report describes a challenging case of an elderly patient with EAS who presented with refractory hypokalemia. Her clinical condition deteriorated rapidly in the absence of surgical intervention.
Case Report
A 74-year-old White woman was brought to the Emergency Department from her nephrologist’s office with a chief concern of persistent anasarca and recurrent hypokalemia of 1-month duration. In addition, she reported generalized weakness and recurrent mechanical falls in the preceding 3 months. Before presentation in March 2021, she had a medical history of type 2 diabetes, chronic kidney disease stage 3b, atrial fibrillation on chronic anticoagulation, heart failure with reduced ejection fraction (EF 35–40%), hypothyroidism, hypertension, and hyperlipidemia. Home medications included diltiazem, apixaban, insulin glargine, levothyroxine, simvastatin, carvedilol, glimepiride, sacubitril, valsartan, and furosemide.
On presentation, she was hemodynamically stable with temperature 36.5°C, heart rate 67 beats per min, blood pressure 139/57 mmHg, respiratory rate 20 per min, and saturation 98% on 2 L oxygen supplementation. Her height was 162.6 cm, and weight was 80.88 kg, with a body mass index (BMI) of 30.6 kg/m2. A physical exam showed central obesity, bruising in extremities, generalized facial swelling mainly in the periorbital region, severe pitting edema in bilateral lower extremities, and moderate pitting edema in bilateral upper extremities. A laboratory workup revealed serum potassium 2.4 mmol/L (3.6–5.2 mmol/L), serum sodium 148 mmol/L (133–144 mmol/L), and eGFR 31.5 mL/min/1.73 m2. Arterial blood gas analysis showed pH 7.6, PaCO2 48.9 mmHg (35.0–45.0 mmHg), and serum bicarbonate 32 mmol/L (22–29 mmol/L), which was consistent with primary metabolic alkalosis, appropriately compensated by respiratory acidosis. Due to concerns of loop diuretic-induced hypokalemia, she was started on spironolactone and potassium replacement. However, potassium levels persistently remained in the low range of 2–3.5 mmol/L (3.6–5.2 mmol/L) despite confirming compliance to medications and adequate up-titration in the dose of spironolactone and potassium chloride. Hence, the workup for the secondary cause of persistent hypokalemia was pursued.
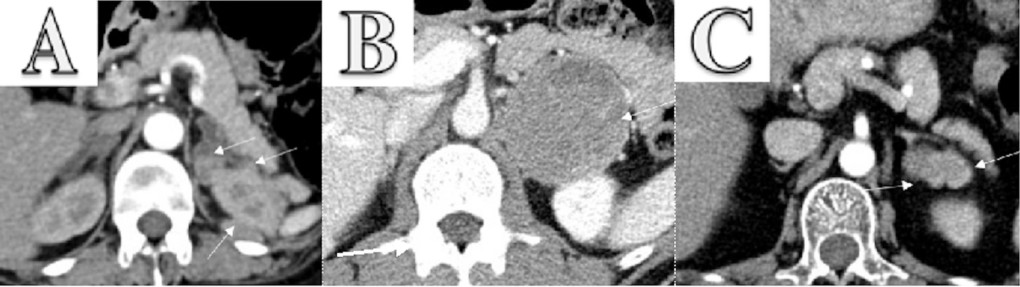
Hormonal evaluation revealed plasma aldosterone concentration (PAC) <1.0 ng/dL, plasma renin activity (PRA) 0.568 ng/mL/h (0.167–5.380 ng/mL/h), 24-h urine free cortisol (UFC) 357 mg/24h (6–42 mg/24h), ACTH 174 pg/mL, and DHEA-S 353 ug/dL (20.4–186.6 ug/dL). ACTH levels on 2 repeat testings were 229 pg/mL and 342 pg/mL. The rest of the laboratory workup is summarized in Table 1. Considering elevated ACTH and 24-h UFC, a preliminary diagnosis of ACTH-dependent Cushing syndrome was made. An 8-mg dexamethasone suppression test revealed non-suppressed cortisol of 62.99 ug/dL along with dexamethasone 4050 ng/dL (1600–2850 ng/dL). A pituitary MRI was unremarkable for any focal lesion suggesting a diagnosis of ACTH-dependent Cushing’s syndrome secondary to an ectopic source. Imaging studies were then performed to determine the source. A CT scan of the chest and abdomen revealed adenomatous thickening with nodularity of bilateral adrenal glands, and a 1.4-cm nodule in the right middle lobe (Figure 1A, 1B). FDG-PET/CT showed severe bilateral enlargement of the adrenal glands with severe hyper-metabolic uptake (mSUV 9.2 and 9.1 for left and right adrenal glands, respectively) (Figure 2A). The uptake of the right lung nodule on PET/CT was 1.4 mSUV (Figure 2B).

CT chest, abdomen, and pelvis w/o contrast showed bilateral enlargement of adrenal glands (A, red arrows) and a 1.4-cm nodule in the right middle lobe of the lung (B, blue arrow).

Whole-body PET/CT following intravenous injection of 40 mCi FDG showed diffuse enlargement of the bilateral adrenal glands with mSUV of 9.2 on the left and 9.1 on the right adrenal gland, respectively (A, red arrows) and low-grade activity with an MSUV of 1.4 in right lung nodule (B, blue arrow).
Table 1.
Laboratory on initial presentation.
| Laboratory test | Level | Reference range |
|---|---|---|
| WBCs | 7.8 k/uL | 3.7–10.3 k/uL |
| RBCs | 3.05 M/mL | 3.–5.2 M/mL |
| Hemoglobin | 9.6 g/dL | 11.2–15.7 g/dL |
| Hematocrit | 27.3% | 34–45% |
| Platelets | 98 k/mL | 155–369 k/mL |
| MCV | 89.7 fl | 78.2–101.8 fl |
| MCH | 31.5 pg | 26.4–33.3 pg |
| MCHC | 35.2 g/dL | 32.5–35.3 g/dL |
| RDW | 15.8% | 10.1–16.2% |
| Glucose | 73 mg/dL | 74–90 mg/dL |
| Sodium | 148 mmol/L | 136–145 mmol/L |
| Potassium | 2.4 mmol/L | 3.7–4.8 mmol/L |
| Bicarbonate | 32 mmol/L | 22–29 mmol/L |
| Chloride | 108 mmol/L | 97–107 mmol/L |
| Calcium | 7.0 mg/dL | 8.9–10.2 mg/dL |
| Magnesium | 1.7 mg/dL | 1.7–2.4 mg/dL |
| Phosphorus | 2.3 mg/dL | 2.5–4.9 mg/dL |
| Albumin | 2.4 g/dL | 3.3–4.6 g/dL |
| Blood urea nitrogen | 41 mg/dL | 0–30 ng/dL |
| Creatinine | 1.60 mg/dL | 0.60–1.10 mg/dL |
| Estimated GFR | 31.5 mL/min/1.73m2 | >60 mL/min/1.73 m2 |
| Aspartate transaminase | 42 U/L | 9–36 U/L |
| Alanine transaminase | 67 U/L | 8–33 U/L |
| Alkaline phosphatase | 90 U/L | 46–142 U/L |
| Total protein | 4.8 g/dL | 6.3–7.9 g/dL |
| Arterial blood gas analysis | ||
| PaCO2 | 48.9 mmHg | 35.0–45.0 mmHg |
| PaO2 | 63.1 mmHg | 85.0–100.0 mmHg |
| %SAT | 92.8% | 93.0–97.0 |
| HCO3 | 47.8 mm/L | 20.0–26.0 mm/L |
| Base excess | 26.3 mm/L | <2.0 mm/L |
| pH | 7.599 | 7.350–7.450 |
| Adrenocorticotropic hormone (ACTH) | 174, 229 and 342 pg/mL | 15–65 pg/mL |
| Urine free cortisol, 24 h | 357 ug/24 hr | 6–42 mg/24 hr |
| 8: 00 AM cortisol following 8 mg dexamethasone (4×2 mg doses) previous day | 62.99 mg/dL | |
| 8: 00 AM dexamethasone following 8 mg dexamethasone (4×2 mg doses) previous day | 4050 ng/dL | 1600–2850 ng/dL |
Based on unsuppressed cortisol following an 8-mg dexamethasone suppression test, negative pituitary MRI, and 1.4-cm lung nodule, we diagnosed ACTH-dependent CS secondary to an ectopic source, most likely from the 1.4-cm lung nodule. While awaiting localization studies, within 3 months of initial presentation, she had 2 hospitalizations, one in May 2021 for acute anemia secondary to bleeding peptic ulcer disease (PUD) requiring endoscopic clipping of the bleeding ulcer, and another in June 2021 for acute on chronic congestive heart failure. The patient’s overall condition continued to deteriorate, and she became progressively weak and wheelchair-bound. A 68-Ga-DOTATATE was planned to establish the source of ectopic ACTH definitively; however, she developed a left hip fracture in July 2021 and could not present for follow-up care. Therefore, she was started on Mifepristone until curative surgery. However, considering the patient’s advanced comorbid conditions, the increased burden of the patient’s health care needs on her elderly husband, and the inability of other family members to provide necessary healthcare-related support, palliative care was pursued. In August 2021, she developed a sacral decubitus ulcer and community-acquired pneumonia. However, she was still alive while receiving palliative care in a nursing home until September 2021.
Discussion
Ectopic ACTH syndrome (EAS) is defined as secretion of ACTH from an extra-pituitary source and is the cause of Cushing’s syndrome (CS) in approximately 4–5% of cases [3,4]. Clinical features of EAS depend on the rate and amount of ACTH production [12]. Among all forms of Cushing’s (excluding adrenal cortical carcinoma), EAS has the worst outcome, with one of the most extensive combined UK & Athens study demonstrating a 5-year survival rate of 77.6%. Compared to Cushing’s disease (CD), patients with EAS have severe and excessive production of ACTH, resulting in highly elevated cortisol levels. This leads to hypokalemia, metabolic alkalosis, worsening glycemia, hypertension, psychosis, and infections. Metabolic alkalosis and hypokalemia are the 2 most common acid-base and electrolyte abnormalities associated with glucocorticoid excess among these patients. Studies have shown that hypokalemia is seen in up to 90% of patients with EAS. Although hypertension and hypokalemia are often attributed to primary hyperaldosteronism, other causes should be sought. Under normal circumstances, the mineralocorticoid effect of cortisol is insignificant due to local conversion to cortisone by the action of 11 beta-hydroxysteroid dehydrogenase. Excessive cortisol in patients with EAS saturates the action of 11 beta-hydroxysteroid dehydrogenase and leads to the appearance of mineralocorticoid action of cortisol [6]. In our patient, the initial treatment of hypokalemia was unsatisfactory, so additional endocrine workup was pursued. Elevated urinary cortisol excretion, plasma ACTH levels, unsuppressed cortisol following 8 mg dexamethasone, and lung mass on CT scan strongly suggested that the clinical symptoms were due to EAS. Unfortunately, despite diagnosing the underlying condition contributing to the patient’s symptoms, her clinical condition rapidly deteriorated without surgical treatment.
Various factors resulted in delayed diagnosis in our patient. First, the patient sought medical care only 3 months after symptom onset. Second, furosemide, a medication commonly used to treat patients with HFrEF, is a frequent culprit of hypokalemia and often is treated with adequate potassium supplementation. Third, multiple hospitalizations resulted in delays in the proper endocrine workup necessary for establishing hypercortisolism. Fourth, localization of the ectopic source requires advanced imaging studies, which are only available in a few tertiary care centers. Fifth, even after tumor localization with PET/CT scan, there is still a need for a more definitive localization study using Ga-DOTATATE scan, which has a higher specificity. However, it was unavailable in our institution and was only available in a few tertiary care centers, with the nearest center being 2.5 h away. Sixth, the impact of the COVID-19 pandemic also played a critical role in promptly providing critical care necessary to the patient. In addition to those, the social situation of our patient also played an essential role in contributing to delays in diagnosis.
It is well recognized that EAS is associated with various malignancies, mostly of neuroendocrine origin. The most common location of these tumors was found to be the lung (55.3%), followed by the pancreas (8.5%), mediastinum-thymus (7.9%), adrenal glands (6.4%), and gastrointestinal tract (5.4%) [9]. Prompt surgical removal of ectopic ACTH-secreting tumors is the mainstay of therapy in patients with EAS [13]. However, localization of such tumors with conventional therapy is often challenging as the sensitivity to localize the tumor is 50–60% for conventional imaging such as CT, MRI, and FDG-PET [9]. In a study by Isidori et al, nuclear imaging improved the sensitivity of conventional radiological imaging [9]. Moreover, newer imaging technologies using somatostatin receptor (SSTR) analogs such as 68Ga-DOTATATE PET/CT further improve the ability to localize the tumor. 68Ga-DOTATATE PET/CT, approved in 2016 by the Federal Drug Administration (FDA) for imaging well-differentiated NETs, has a high sensitivity (88–93%) and specificity (88–95%) to diagnose carcinoid tumor [14]; however, a systematic review reported a significantly lower sensitivity (76.1%) of 68Ga-DOTATATE PET/CT to diagnose EAS [15].
Once localized, the optimal management of EAS is surgical re-section of the causative tumor, which is often curative. However, until curative surgery is done, patients should be medically managed. Drugs used to reduce cortisol levels include ketoconazole, mitotane, and metyrapone [16, 17]. These are oral medications and decrease cortisol synthesis by inhibiting adrenal enzymes [17]. Etomidate is the only intravenous drug that immediately reduces adrenal steroid production and can be used when acute reduction in cortisol production is desired [16].
Medical management requires frequent monitoring of cortisol levels and titration of dose to achieve low serum and urine cortisol levels. Mifepristone, an anti-progesterone at a higher dose, works as a glucocorticoid receptor antagonist and can be used to block the action of cortisol. Its use results in variable levels of ACTH and cortisol levels in patients with EAS. Hence, hormonal measurement cannot be used to judge therapeutic response, and clinical improvement is the goal of treatment [18]. Drugs inhibiting ACTH secretion by NETs such as kinase inhibitors (vandetanib, sorafenib, or sunitinib) are effective in treating EAS secondary to medullary thyroid cancer [19]. Somatostatin analogs such as octreotide and lanreotide have demonstrated short- and medium-term efficacy in a few EAS patients; however, a few patients failed to improve, necessitating the use of more effective treatment options [19,20]. Hence, they are not considered a first-line drug as monotherapy and should be used in combination with other agents, or as anti-tumoral therapy in non-excisable metastatic well-differentiated NETs [19,20]. Cabergoline, a dopamine agonist, has been used with variable therapeutic effects in a few patients [19]. In 1 patient, the use of combination therapy using Mifepristone and a long-acting octreotide significantly improved EAS [21]. In our patient, we initiated Mifepristone to reduce the burden associated with frequent biochemical monitoring and planned 68Ga-DOTATATE PET/CT to localize the tumor; however, further diagnostic and therapeutic approaches could not be further undertaken per family wishes.
Conclusions
EAS can present with refractory hypokalemia, especially in patients who are already at risk of developing hypokalemia. Diagnosis of EAS is often challenging and requires a multidisciplinary approach. Localization of source of EAS should be done using nuclear imaging, preferably using SSTR analogs, when available. Urgent surgical evaluation remains the mainstay of treatment following tumor localization and can result in a cure. EAS is a rapidly progressive and life-threatening situation that can be fatal if diagnosis or timely intervention is delayed.
Abbreviations
| ACTH | adrenocorticotropic hormone; |
| CS | Cushing’s syndrome; |
| CT | computed tomography; |
| EAS | ec-topic ACTH syndrome; |
| MRI | magnetic resonance imaging; |
| FDG/PET | 18-F-fluorodeoxyglucose positron emission tomography; |
| NET | neuroendocrine tumors; |
| SSTR | somatostatin receptor; |
| EF | ejection fraction; |
| PAC | plasma aldosterone concentration; |
| PRA | plasma renin activity; |
| UFC | urine free cortisol; |
| DHEA-S | dehydroepiandrosterone sulfate; |
| 68-Ga-DOTATATE | Gallium 68 (68Ga) 1,4,7,10-tetraazacyclododecane-1,4,7,10-tet-raacetic acid (DOTA)-octreotate; |
| PUD | peptic ulcer disease |
Adrenal Surgery: One Patient's Experiences
in Cushing's Basics
Posted
From another patient...
Adapted from a thread on the message boards.
I'm going to try to keep all of my post-op BLA updates in this thread. I am hoping it will eventually show positive progression and be a realistic and inspirational thread for others.
Today I am two weeks post-op BLA. So far, no scares. I am on 30/20/20 of hydrocortisone and weaning by 10 mg every four days. I am sleeping a good bit during the day and resting a lot to get my strength back. If I am upright too long my abdominal area aches and I get fatigued, sometimes it still aches even if I am not upright. My nighttime sleep has been good. I'm waking up only 1-2 times to go to the bathroom (I think the meds are making my bladder more active than normal), but otherwise am sleeping through the night which is a huge change from Cushing's. I am hoping this is due to being Cushing's-free rather than just due to the pain meds I am taking right now. We'll see if this lasts as I drop the pain meds and hopefully the nighttime urination will let up as the hydro levels drop.
Also, and I don't think its my imagination, but some of my stretch marks are getting lighter. In particular, the ones that formed on my legs after my pit surgery. This is a positive sign! I showed my mom and hubbie and they could both see the change too. Unfortunately, my hump is bigger right now than pre-BLA and my cheeks are still pretty red, but I bet this will change as I wean down.
No weight changes as of yet, but not expecting any because I am still on such a high dose of hydrocortisone. I was 198 the day of the BLA, which was about 15 pounds heavier than the day of my pit surgery seven months ago. For the first week and a half after the BLA I was really, really bloated - and it was all in the stomach area. Most of this bloating has gone down in the past two days.
I've watched my calorie intake throughout the battle with Cushing's but I started a food journal yesterday just to make sure I am keeping myself in check. I'm eating 1500 calories a day. I noticed right away that I haven't even been eating that much on a normal basis because I actually had to eat more than normal to meet the 1500 calories. So that's also a good sign that watching my food intake won't be a big change in order to help the weight to come off.
So that's really the main things happening right now. Just taking things slow and steady and trying to have realistic expectations!
I had my six week post op appointment in Seattle last week. My weight is actually up (204, I was so bummed that I went over 200). But Dr. L said not to worry, that its normal to gain weight during the weaning process. I am still on a 1500 calorie a day diet and will stay there until I start to see weight loss and then I'll reassess calories then. I was advised that weight will probably start to fall off when I'm six months out from surgery, so I am trying not to focus too much on it or get discouraged.
My nighttime sleep is weird right now. I'm not waking up all night long like I was before the BLA, but I can't fall asleep at night either. I lay awake until 1 or 2 am. On the flip side, I am waking up at a normal hour - 7 am.
I just started weaning to 20/5 of hydrocortisone. It is pretty rough. The wean from 20/10/10 to 20/10 was hard, but this is even harder. Feels like the flu, achey all over, headaches, sleeping all day (which probably doesn't help me fall asleep at a good time at night!). I have realized that I must take the wean really slowly now. The goal is to get to 20 or maybe just a little less and hopefully that dose will work for me.
In other news, I got the path report back on my adrenals - my adrenals combined weighed in at 30 gm (normal combined weight of adrenals should be between 8-12 gm). The left one was twice the weight of the right one, and they had "subtle vague expansion" and "microscopic nodularity" suggestive of adrenocortical hyperplasia.
So I am feeling very validated at this point and I know I made the right decision to have the BLA.
I'm just past the 3 month post-op anniversary. Some things are better and others are still the same. But more positive changes than anything.
We'll get the negative overwith first - my stretch marks did an about-face and actually got a lot worse about a week after I got down to my physiological dose (20mg). Dr. L said not to worry, they're just showing up now due to past cortisol exposure. Still, they're pretty bad. So I was disappointed in that. My period still has not come back since I had the pit surgery. All my hormones are fine except the progesterone, but progesterone supplements are not helping. We're taking a wait and see approach to give my body some time to get over the shock of two major surgeries.
Other than the stretch marks, the other Cushing's symptoms are slooooowly getting better. I am sleeping pretty well now, able to fall asleep in the evening and sleep until 5:30 or 6 am until waking up. Its a lot better than waking up at 3 am every night for sure. My hump looks a little smaller (I think). My cheeks are still red, but my face is maybe slightly slimmer (I think). I've lost six pounds (with 80 more to lose), but am still heavier than I was the day of my BLA. Although my stomach doesn't pooch out so much anymore, so I look less pregnant. My hair has stopped falling out.
I have been working out for a few weeks now and my strength is really starting to improve. Walking is very good for me. I'm eating about 1200 calories a day and dropping down this low seemed to jumpstart some weight loss. I am hoping it continues. I'm certainly doing nothing food-wise to keep the weight from coming off.
I was tested for insulin resistance and any thyroid problems - everything came back normal. My ACTH was super low when it was last checked - came back at 3. (yay!!!)
I went back to work 80% time this week. I'm trying to work short days but my work is very demanding so I will probably have to end up working 4 days a week and taking off one day a week to rest. I am very tired at the end of the work day. Exposure to stress is also very hard on my body - the stress I have encountered this week has caused nausea, diarrhea and one time I had to take straight to the bed and lay down all evening. Right now I feel like I am not as sharp and "on the ball" as I used to be.
My sinus infections from the pit surgery keep continuing about every 6-8 weeks. I've probably had at least 4-6 sinus infections since March. At the last visit to the ENT doc, she said she thinks I have a deviated septum from the pit surgery and may need surgery to correct it. I have a CT scan on Tuesday so hopefully we'll know more soon on whether I am having another surgery.
But overall, I just feel better. The Cushing's symptoms are slowly fading, but at least we're going in the right direction. I am trying to be patient, and trying to remain motivated. I have to admit I am becoming very impatient for the weight to come off and still harbor fears that it won't. I am considering throwing my scale in the spare bathroom and forgetting its existence for a while.
I hope my next update will have tons more good things to share.
So I am 6 months post BLA today. Yay! This is the magical date - things are supposed to start changing more quickly after passing this milestone. Here's the stats so far:
20 mg hydrocortisone per day
0.1 mg florinef per day
Had thyroid checked in January - fine
Had glucose tolerance test in January - fine, no insulin resistance
Dr. L didn't think I had GH issues at my 3 month post op appointment
Estrogen and all other female hormones fine except progesterone, taking prometrium to try to induce period with no success so far
I started losing weight at the end of January through mid March. I lost 10 pounds. But now, I haven't lost any weight in over a month and I've actually regained two pounds. I am exactly what I weighed the day of my BLA now.
I've been working out 90 minutes 4-5 days a week (elliptical machine and weights). I'm eating net 1200 calories a day (which means I am actually eating more than 1200 because of all the exercise I am doing) and very closely tracking calories on livestrong.com.
I have to say I am very frustrated at this point because I'm working so hard and not losing weight. I'm going to bring this up with Dr. L at our six month post op appointment. If some other BLArs could chime in and tell me what to expect for the next six months, I would greatly appreciate it. Just starting to get a little nervous here.
As far as the Cushing's goes, I have more energy and I am sleeping better. Most nights I sleep through the night and if I wake up, its only once and closer to 6 am than 3 am like it used to be. Hump is still there, hasn't gone away but is a tad smaller. Hair stopped falling out a while ago and has stayed just fine, no relapse.
The stretch marks (which had gotten worse after the BLA) are getting much better, at least the ones on my legs. Those are noticeably better. I've gotten comments that my face is slimmer and I look like I've lost weight. I've gone down from third trimester maternity pants to second trimester pants. That is some progress because I look less pregnant.
Since my last update, I have had three severe episodes of AI. All occurred late at night following a week of being pushed beyond my medical restrictions at work. 32 hours a week seems to be a good balance though, more than that causes me to be really tired and at risk for AI.
I'll close out with a great accomplishment story. Hubbie and I went on a cruise to Mexico and Belize. I was able to do a hike through the jungle (which was relatively level, for a jungle). But the best part was when we got to a clearing and saw the Mayan temples. You could climb one that was about 45 meters high with very steep stairs to the top. Of course my hubbie was the first in the group to take off up the temple. The stairs were so steep they had a rope that came from the top all the way to the bottom to pull on to help yourself get up. This was the type of thing that, before Cushing's, I would have been right there with my husband.
He was about halfway to the top when I said, "Heck, I'm going too." Probably shouldn't have, but I took off up the temple stairs after him. I climbed up and up in the Belize heat and made it to the top. The view was rewarding, but the greater reward was that I could DO it. I was getting part of my life back - the adventurous, hiking, exploring, running-being-free part.
That part was the best.
I am weight training, 4x a week for 20-25 minutes per session, on machines, not free weights. I want to make sure I'm not getting the wrong form. I am pushing myself, sometimes only able to do five reps at a time because of the heaviness of the weight. I do a total of 3 sets of 10 reps per exercise. I'm doing upper body and lower body on different days, so 2 days a week of upper and 2 days of lower, never back to back.
My diet is good. Short of starting to cut out food groups altogether, there's not much else I can do. I eat either whole grain cereal and skim milk or two boiled eggs and skim milk for breakfast. My mid-morning snack is fruit - usually a cup of red grapes or an apple. Lunch is a salad with grilled chicken or a Lean Cuisine or Smart Ones that has fish as the main entree - nothing over 300 calories. Mid afternoon before working out I have fat free yogurt or 30 almonds. Dinner is normally something like stuffed green peppers or chicken fajitas - usually about 500-600 calories.
I weigh/measure just about everything . . .
I'm 7 months 9 days post op today. The weight has changed a little, but only a little. At least its going down and not up, but I admit I am frustrated with my progress. I told Dr. L about my intense working out and dieting and he suggested I wean some more. So I weaned to 17.5 mg of hydro first and then down to 15 mg. I've been at 15 mg for 3 weeks now. The past week I started to see some progress - I lost 2.5 pounds this past week, so now for a weight loss total of 12 pounds since January. This is in conjunction with a 1200 calorie a day diet. I've now gone to a combination Zone diet (30 protein, 30 fat and 40 carbohydrates) and sort of low glycemic index - just as little sugar as possible. So I am eating a lot of bran, salads, chicken and fish. I've instituted a "salad for dinner two times a week" rule at home, which my lovely, Southern-food loving husband has generously agreed to go along with.
May was not as good a workout month as March and April. However, we did a one week vacation with LOTS of activity - hiking every other day for 2-3 miles, and we did a 14.5 mile bike ride at the end - it was mostly flat, but still! That was a long way and I was so proud of myself when I finished it. It was a struggle, but I did it.
I also got my period for the first time in over a year in May. I wonder if it is related to weaning to 15 mg? We will see if it comes back in June . . . .
Other things have gotten much better - sleeping well through the night, feeling better in general. My hair was much better until the past two weeks or so when I've seen more of it coming out in the shower than normal (what is that all about?!?!?) but not falling out on a regular basis like it was with Cushing's there at the end.
I am losing some inches for sure and I don't look as pregnant as I used to, I was able to drop from my maternity black dress pants to a size 18 pants (although the legs are still huge). I am still in my maternity jeans but I did go from trimester three to trimester two! I picked up prescriptions at the pharmacy today and my pharmacist said "You are looking great!" So that was nice to hear
So all in all, very very slow but seeing some progress now. I think its going to be a very long process with lots of hard work and healthy eating. It may take some more weans too, depending on whether I hit a wall again.
I know you and a lot of other BLA-ers are struggling right now. Its hard. I feel good right now because the scale went down this week and I've seen some physical changes in how my clothes are fitting. I know its depressing when you are not seeing that. But for you and everyone else, just hang in there. Do as much physical activity as you can, and at least control your diet, because that is in your control. I know we're told the weight is supposed to come off on its own but I can tell a difference when I'm eating right and when I'm not. At least for me, I think it does help with the weight loss. At least psychologically I know I'm doing everything I can to make it come off.
By way of a mini update, I have lost another 2 pounds since I posted three days ago. This is getting exciting! And its not just water, you know the size 18 blank dress pants I just talked about in my last post? They are now TOO BIG!!! A friend of mine hadn't seen me in two weeks and she was shocked today just to see the changes that have happened in two weeks. It really is noticeable.
Ok, hope I am not jinxing myself. When I update again in a few weeks hopefully I can report a very large weight loss and even more changes!
So, today I am 9 months post-op BLA. Its been almost two months since my last update. There's been a lot of developments:
- In July, I got the results of my bone density scan: I have osteopenia and a severe vitamin D deficiency. I am now on 1200 mg of calcium a day and 50,000 IUs of Vitamin D a week.
- Hair is doing great! Not falling out, shiny, less frizzy.
- Energy is ok. Work is wearing me out, still working me beyond my medical restrictions, but I am supposed to be moving into a new job at the end of next month that will hopefully take care of some of that. I tend to get sleepy during the day and stressful days make me weak. I've also started waking up in the middle of the night again (NO!!! Why is this happening?!?!?) and there for a while I was waking up to pee in the middle of the night again. I wish that would stop because I was enjoying sleeping all the way to the morning.
- Stamina is great. I did a two-hour workout last week (weights and cardio) that was intense and awesome. I was so proud when I was done. I am considering returning to kickboxing in a few months if my Vitamin D levels go up and I have some confidence that my bones have gotten stronger.
- The weight is stalled out. I have lost 16 pounds now, but I haven't lost a pound since mid-June. I weaned to 10 mg of hydro about three weeks ago and no results even with doing that. I don't feel comfortable going any lower than that. Still at 1200 calories a day and low glycemic diet, heavy on protein, very little to no bread or cereal products. Husband and I met with reproductive endo here in Atlanta today (who I love!) and he expressed concern. My thyroid and insulin resistance tests are normal but he's putting me on some Synthroid and Glucophage and some Prometrium. When I got my period in May the weight was just falling off . . . he thinks its PCOS-like issues and this combo of meds might help. So we're going to try that and see how it goes.
- Stretch marks are much, much better - I noticed a marked difference after I weaned to 10 mg hydro. BLA scars are lightening too, especially with help of some new special cream from my dermotologist.
- Haven't gotten my period again Boo. Hopefully the above cocktail will help with that.
Boo. Hopefully the above cocktail will help with that.
- Had lasik surgery!!!! I love it. I did stress dose 30 mg extra for that. I did just fine.
So, positives yes but still very bummed about the struggles with the weight. I am hoping the new medicines will give me some results. I also feel like I've become more emotional lately because I'm tired of eating lettuce, spinach and egg whites (yes, that makes up a large portion of my diet) and working out and getting no relief. I hate being emotional and moody and feeling like I just can't take it anymore. So I certainly do have those days. But thankfully they are just days - usually just one - and it passess and the next day I'm back in the battle. Because really - what else can you do?
I'm 10 months post-BLA today. Unfortunately, this update is not going to be as positive as some of my past updates.
The weight loss stands at 20 lbs now. I did start on Metformin and Synthroid at the beginning of August. I lost five pounds right away the first week, and then the weight loss stopped and I have gained back one pound. Nothing else has happened since then (despite doubling the dose of Metformin).
I can't deny that I have become extremely depressed. Its been building for several months now. Its not just having the extra weight, but the weight keeping me from what I want to do - principally, have a baby. I've just lost interest in so many things and I am very down, despite the progress I have made in other areas of recovery.
I have discussed this with both Dr. L and my reproductive endo. I am going to Seattle in two weeks and we're doing a round of labs and a growth hormone stim test. GH deficiency would explain a lot of things - the large amount of weight around the middle, the Cushie-like shape I still have. I still have a bit of a hump too.
My reproductive endo is re-testing all my thyroid hormones, estrogen, progesterone and a few others soon as well.
I am beginning to suspect I have slowly been becoming hypo-pit. Or perhaps hypo-pit in an intermittent way. I have no menstrual cycle anymore. I have ostepenia. I have energy to do stuff but then I get exhausted and sometimes it takes me days to recover. I have hot flashes, memory issues, loss of libido and insulin resistance. And, again, super slow weight loss that seems to go up every time I eat anything other than raw vegetables. I also have on and off DI.
So, I guess I am just at the end of my rope. I hope that someone can fix me. Because something is still clearly wrong.
I'm now 10 1/2 months post-op BLA. I just completed a visit to Dr. L in Seattle. I did the GH stim test and labs for thyroid, ACTH and some other things.
As I suspected, I do have some continuing issues - I am severely GH deficient. I didn't stim above 0.9 during the entire stim test. I'll be starting on GH as soon as possible.
My thyroid numbers are all in the normal range but they are low normal. We're upping the Synthroid to 125 mg per day.
My MRI was clear - no new tumor (yay!) and my ACTH was 40. So that is all good. I feel hopeful that I am doing good in some areas and now we have identified the areas that are causing me problems.
I also had estrogen, FSH and LH tested today. I am hoping to find out if I am deficient there even though I haven't been in the past - I have a suspicion the estrogen may be low now.
So, we'll see where we stand in a few months when this medicine has had some time to kick in.
Today is the one year anniversary of my BLA. I am doing well. I'll update here and post a separate 1 year post-op BLA thread so those who don't follow here can be encouraged by my, dare I say it, success story?
The past month and a half I have seen some significant improvement. Here's the breakdown:
Medicine every day:
12.5 mg of hydro (all taken in the morning)
0.1 mg florinef
1500 mg Metformin at night
125 mg of levothyroxine
Calcium pill and daily multivitamin
Progestrone pills on days 1-10 of each month
To start 0.2 mg of Genotropin in next few weeks
Energy: The thyroid medicine has helped a lot with energy. My thyroid numbers were all normal but just a bit on the low normal, so the docs didn't think I needed meds. But I did, it has helped a lot. I am still tired but I am a lot better than I was. My GH is supposed to arrive today (yay!) so that should also help me on my path to recovery.
Weight loss: I haven't really lost weight in the past few weeks but inches, oh my! I have lost inches. I have gotten tons of comments from friends, family, coworkers, etc on the change all over - face, body, etc. I am now down to a size 14. That is down from being mostly in maternity clothes and barely squeezing into a few size 18 pants a year ago. No more maternity clothes for me (for now!). Its so nice to be shopping in the regular clothes again. I have gone a bit crazy buying some new things - skinny jeans, sweaters, ballet flats, boots. I am all decked out for fall in the latest styles. It feels so good to be stylish and to have choices again.
The pregnancy look is gone. No more comments on when I am due or what sex the baby is. That is an awesome feeling. I've lost 20 solid pounds, some days a little more but it seems to always go back to that 20 number. I am trying not to weigh too much until the GH has a chance to start working.
Stretch marks: My stetch marks have really done some fading. Somedays they are more noticeable than others, but they are so so so much lighter than they were.
Hump: My hump is much smaller - its barely there at all now, I probably see it only because I am paranoid. But I have no issue wearing tank tops or anything that shows the back of my neck.
Hair: My hair has grown long and thicker than it used to be. Much less oily! I don't have to wash it every single day now. I can put it in a ponytail on the weekends and it looks cute and not greasy.
Sleep: I am sleeping great. I sleep all through the night and don't wake up anymore. That has become very consistent, which is a wonderful thing. I still feel tired though because of the GH but hopefully that will improve.
Activities: I work 32 hours a week. This works well, it gives me an extra day a week to rest and recover from the work week. I exercise often, I went back to kickboxing this month which is kicking my butt but I need to rebuild muscle. Its also nice to get back to your old hobbies.
Attention and Memory: This isn't always as great, hoping the GH will help. I don't focus as well as I used to or catch spelling details at work like I used to. Sometimes my memory is sharp and other times I forget something someone just told me or how to spell a word or the names of objects (or even people at times). Again, hoping GH will help here.
Female stuff: No period still, progesterone is not really helping. I think if the GH doesn't help here, we're going to move on to estrogen therapy in a few months. We're hoping to start trying for a baby at the end of next year.
Other health issues: High blood pressure went away immediately after the BLA. I never did have a blood sugar problem so no issues there. I do have osteopenia which we are working on with more calcium and Vitamin D and weightlifting exercies. Again, hoping GH will help here.
Emotional: Really doing much better. I was getting really depressed for a while there, about 7-10 months post op. Even though I thought my expectations of recovery were reasonable, I was frustrated with my progress. I had hoped to lose more like 30 or 40 pounds in my first year. But, I found out I had other issues (thyroid and Gh deficiency) that were messing with that goal. So I only got halfway there but it wasn't my fault. I also thought the weight would "fall off" more than it has, but it hasn't. I've had to diet and exercise hard for every pound lost. Don't know if that's normal or if its just me or because of the thyroid/Gh issues we are still working on. But it helped me to know that at least there was a medical reason for my frustrations!
But overall I am really doing well. No one who meets me for the first time has any idea that I've been sick. I recently started a new position with my same employer (which has been going well) and I met my new team and everyone commented on my "glowing skin" and "happy nature." I have no regrets about the BLA.
My advice to anyone considering it or just having had the BLA is: patience, patience, patience. Realistic expecatations. Then, hard work on controlling your diet and being physically fit. Do everything you can towards getting better, and then if time and hard work don't pay off, don't hesitate to detail your hard work and patience to your doctor and tell them to find out what else is holding you back.
I hope this is an inspiration to anyone out there who is struggling right now.
Wow, I am way overdue for an update! I've been out working, having fun and living my life!
Its been a little over three months since my last post here. I am now 1 year, three months and ten days post op BLA. Here's the breakdown of where I am now:
Medicine every day:
12.5 mg of hydro (all taken in the morning)
0.1 mg florinef
1500 mg Metformin at night
150 mg of levothyroxine
Calcium pill and daily multivitamin
Progestrone pills on days 1-10 of each month
0.2 mg of Genotropin 7 days a week (started in November)
Energy: I am doing great here. I am tired sometimes and traveling or working long hours wears me out, but I have limited that in my life with my new job. At my new job I am working full time now, 40 hours a week. I also work out 5-6 days a week now at very energetic things like kickboxing (with punching bags) or the elliptical machine.
Weight loss: I didn't lose anything between my last post and the end of December. When my thyroid medicine was raised to 150 mg at Christmas, combined with continuing my workout and diet, I really started to see results. I have lost 10 lbs since then, for a total of 31 lbs now. Still, I am not losing at the rate I should be for the math of the intake/output of my diet and working out. We are working on that, possibly some more meds to come soon. But it is much, much improved! I have about 35 pounds to go until I am at a good weight for me. Ideally I'd like to lose 45 more but 35 more would be a healthy weight for me.
The best thing I did was have my husband hide the scale. I only weigh every six weeks now. Now I can focus on the process and not focus on how hard it is to get the scale to go down or get depressed when it doesn't budge. I am now in size 12 clothes. I was a 6-8 before Cushing's, sometimes I could wear a 4. I have a few more sizes till I can wear most of the clothes in my closet.
Stretch marks: This is about the same since my last post. My stetch marks are almost all white. Somedays they are more noticeable than others or pinker than usual, but they are so so so much lighter than they were.
Hump: Same as last post - much smaller and hardly there at all.
Hair: Same as last post - doing great.
Sleep: Same as last post - doing great.
Activities: Like I said above, working 40 hours a week, kickboxing probably 3-4 times a week, other days I work out on the elliptical machine and lifting weights. I go walking or hiking with my husband on the weekends if the weather is nice, but this low impact working out didn't do much for the weight loss. The kickboxing has really helped.
Attention and Memory: This is about the same as last time. I can't tell that it has improved all that much. I forget things (like reminding my husband to do something when he has asked me to remind him) all the time.
Female stuff: No period still, had blood drawn for estrogen today. Will see whether I am going on that or not.
Other health issues: High blood pressure went away immediately after the BLA. No return of that, blood pressure is very good. I never did have a blood sugar problem so no issues there. I do have osteopenia which we are working on with more calcium and Vitamin D and weightlifting exercies. Again, hoping GH will help here. My sinus issues have really escalated and just never got better after surgery. I've had a persistent sinus infection for two years. I have mold and some other bacteria in there that countless treatments have not killed. I am having the sinuses washed in a surgery at the end of the month and am now working with an infectious disease doctor to try to kill it. Its too gross to talk about!
Emotional: I am really doing well. The recent weight loss has really pleased me. I don't think I am at the maximum improvement for my weight loss rate yet, but hopefully we are getting there. I am pulling out old clothes I haven't worn in years out of my closet. I now officially weigh less than my husband for the first time in over two years, which is also wonderful.
So, that is about it for now. I will update again when there are more developments!
P.S. - Notable fitness accomplishment! Six weeks ago throughout a kickboxing class I could do about 5 girlie push-ups (on knees). Last night at kickboxing class I did a total of 5 interspaced intervals of 10 for a total of . . . 50 push-ups!
The power of regular exercise and GH unites!
Today is the two year anniversary of my BLA. It is hard to believe that much time has passed. I can say with 100% confidence that I am doing so much better and that the BLA was the right thing for me.
I'll update this along the same lines as my one year update, just in the name of consistency:
Here's the breakdown on my meds:
Medicine every day:
7.5 mg of hydro (all taken in the morning)
0.1 mg florinef
125 mg of levothyroxine
Calcium pill and daily multivitamin
Prenatal vitamin
0.6 mg of Neutropin (next month will be going up to 1 mg Neutropin)
Birth control pills (formerly was taking 0.2 mg estrogen supplement and progesterone on days 1-10 of month)
2 tsp. of Royal Jelly and Bee Pollen in honey daily
Flonase
Energy: The thyoid and GH have helped a lot in this area. I could still use a little help because my GH is still very low, but I really am doing great anyways. Getting the thyroid dose right has been a battle, but I think we finally found the right dose.
Weight loss: I have now lost a total of 34 lbs, down from high of 206 to 172. At 5'5 I am a normal size 12 and its great. I look and feel like a normal person again (my mom even says I am "skinny" but I don't know about that!) I am losing more inches now than I am weight. This is partly due to the need for higher GH, and partly because I am not doing the hard working out and strict dieting because my hubbie and I are working on Baby #1!!! I have fought hard with diet and exercise for every pound lost - nothing has come off easily for me.
So, the pregnancy look may be back in a few months, but this time it will be because I am actually pregnant
Stretch marks: i barely notice them at all now. My BLA surgical incisions have done a great job fading as well. I don't know if a bikini is ever in my future, but if I am in that great shape again I might wear one around family and friends despite the scars.
Hump: Gone
Hair: Doesn't fall out anymore, its grown long and thicker, less oily. I think the prenatal vitamins have helped in that area too.
Sleep: I sleep like a baby every night. I have been for a while. No more waking up, no more problems falling asleep. I do need more sleep than most people, and I am wondering if this is still due to the GH deficiency.
Activities: I work 40 hours a week and have been since probably the beginning of the year. I've been in my new job now for a year and it has been such a blessing. The reduced stress makes it possible for me to work full time.
Attention and Memory: This is the same as last year. It isn't always as great, hoping the GH will help. I don't focus as well as I used to or catch spelling details at work like I used to. Sometimes my memory is sharp and other times I forget something someone just told me or how to spell a word or the names of objects (or even people at times). Again, hoping GH will help here.
Female stuff: I need a combination of estrogen and progesterone in order to have a period. This still does not cause ovulation. So, we are using fertility mediation to induce ovlutation in order to get pregnant.
Other health issues: Same - High blood pressure went away immediately after the BLA. I never did have a blood sugar problem so no issues there. I do have osteopenia which we are working on with more calcium and Vitamin D and weightlifting exercies. Again, hoping GH will help here. I had some problems with my gums recessing and GH and better female hormones have helped there too.
My sinus recovery from the pit surgery has really been hard, perhaps my worst problem of all. I had surgery in April to correct the deviated septum caused by the pit surgery. I have been on and off antibiotics like crazy. I was a habitual Neti-Pot user with no improvement. Finally, I started using those spray irrigation cans twice a day, combined with Flonase to lessen the mucus, and that has helped for the past 8 weeks. I've seen my best improvement since by pit surgery 2.5 years ago. So let's hope that continues.
Emotional: i am really very happy in my life. I am not depressed anymore and so many good things are happening to me. I thought I would have lost more weight by now but solving the GH deficiency has really taken a long time (and its still not resolved yet). Also, its important when using fertility medications to take it easy and not eat a restrictive diet, so I've been focusing more on the things to help us have a baby more than weight loss. I pray we are successful in having kids, and I will get back on the weight loss track after that. But its so positive to shop in normal clothes and not even be considered plus size anymore!
My relationship with my husband is great, unlike so many relationships we pulled together through Cushing's and it made us stronger.
I am still working to have patience in the recovery and just to recognize that it goes on for a long while. I am two years out and things improve all the time. Its just good to be in a place where things are getting better rather than worse, and I can eat a piece of pizza and not gain 5 lbs, and actually be out enjoying life. Hopefully this next year I can tackle motherhood too
So far the BLA hasn't been the doctor's concern at all for getting pregnant. The problem has been the lost pituitary hormones from the pituitary surgery. If I get pregnant, there will be focus on keeping the cortisol levels appropriate, as they rise naturally during pregnancy and my meds will have to do that. But I would guess someone who did not have a BLA and had pit surgery and is still reliant on cortisol replacement would have the same issue.
There is also some focus on cortisol dosage if I have morning sickness in order to avoid AI, but the docs don't seem too concerned and feel confident we can handle it.
PS- this was why I chose the BLA over the second pit surgery, although I lost ovulation with the first pit surgery, so fertility meds were unavoidable.
Wow, I can't believe it, but yesterday was the three month (year!) anniversary of my BLA. I am doing awesome. Honestly, I hardly come on the boards anymore but I am trying to update this thread at least yearly in the hopes that it will help someone. Here is an update on the areas I have traditionally noted:
Here is the breakdown on my meds:
Hydrocortisone: There is controversy here. Technically, I am supposed to be taking 7.5 mg a day as the minimun. But its too much for me. I can live without it. I have gone months living without it. Every now and then if I feel bad I will take 5 mg. The rest tissue testing I have done at Vanderbilt has been negative for rest tissue, but clearly something is going on. I've also lost weight being off of the hydro.
Fludrocortine: Again, I am supposed to be on 0.1 mg a day, but I can live without it. I may need to take a pill once every three or four weeks, but otherwise I am fine right now.
125 mg of levothyroxine
0.6 mg of Nutropin
Calcium, multivitamins
Vaginal progestrone suppositories - these, combined with no hydro, have really helped the weight peel off
Estrogen patch - same, have helped the weight come off, because oral meds interfere with GH
Energy: I am doing great, working 40+ hours a week. Sometimes pain in my knees interferes with my workouts, but otherwise I am doing fine as long as I get 8-9 hours of sleep a night.
Sleep: doing great, fall asleep and usually no waking up.
Weight: Awesome, i made huge strides this year with the change in the manner in which female hormones are put into my body and going off the hydro. I lost 30 lbs this year, and I have now lost 64 of the 66 I gained with Cushing's. I am wearing a size 6 or 8 depending on the store.brand. Before Cushing's it was a 6 or a 4. But after all this, I consider this a huge success story
Hump: still gone, and man, do I have collar bones now!
Hair: still doing great
Stretchmarks: Not very noticeable, and the BLA scars are very faint. A friend of mine (who saw them after surgery) saw them yesterday for the first time in three years and was amazed.
Other health issues: High blood pressure gone, high cholesterol gone, sinus issues are still present but I have now had two sinus surgeries. I may be going into IV antibiotic therapy next.
As far as Baby #1, I had a miscarriage in March but we determined the reason was not Cushing's related and another fixable problem I had. So, hopefully in the future I will get my bundle of joy. I am much happier that I am now at a healthier weight for it (142 lbs at 5'5).
Again, so happy I made this decision. I consider myself fully cured, and I am still losing weight now without much effort. Before this year, I was fighting against unbalanced hormones and while I did lose 34 lbs during that time, it took me two years! This year, only one year and 30 lbs. Balanced hormones are totally necessary, but you also need the proper manner of distribution to your body, and healthy eating and exercise.
I hope this helps someone along their Cushing's journey! There is hope and light at the end of the tunnel.
Time for another update I guess. I am continuing to do really well. I am down to 118lbs at 5'5. I am a size 4, sometimes a size 2. I never thought I would see any of those numbers again, but here I am! I am feeling good in pretty much all respects. The only bad thing is that I seem prone to sports injuries. I don't know if its because I'm post-Cushings or if its just me. I've been in physical therapy twice in the past year now. But I am continuing to be active and have a healthy lifestyle.
I hope everyone is doing well. As always, let me know if you have questions about anything in my journey.
Wow I didn't realize how long it had been since my last update! So much has happened in the last 8 years. I've gotten divorced and since remarried. The biggest update is that I am pregnant from IVF and expecting my first child. There was always a question after my pituitary surgery on whether this would be possible. But I froze my eggs in 2013 and 2014 and finally can say that investment paid off")
The pregnancy has put a lot of stress on my body so I've had to go back on hydrocortisone and fludro. I've been off of both for about ten years now and surviving just on my rest tissue. I've done incredibly well! So far I've only gained a little more than what you are supposed to while pregnant so losing the weight will be my next project once this baby is born. I'm in my third trimester now.
Its been an incredible journey. I remember reading these boards and struggling to find anyone who had had a BLA and then gotten pregnant. I hope my journey will continue to help and inform others.