

MaryO
-
Posts
8,082 -
Joined
-
Last visited
-
Days Won
555
Content Type
Profiles
Forums
Events
Blogs
Gallery
Articles
Media Demo
Store
Posts posted by MaryO
-
-
Abstract
Purpose:
Literature regarding endogenous Cushing syndrome (CS) largely focuses on the challenges of diagnosis, subtyping, and treatment. The enigmatic phenomenon of glucocorticoid withdrawal syndrome (GWS), due to rapid reduction in cortisol exposure following treatment of CS, is less commonly discussed but also difficult to manage. We highlight the clinical approach to navigating patients from GWS and adrenal insufficiency to full hypothalamic-pituitary-adrenal (HPA) axis recovery.
Methods:
We review the literature on the pathogenesis of GWS and its clinical presentation. We provide strategies for glucocorticoid dosing and tapering, HPA axis testing, as well as pharmacotherapy and ancillary treatments for GWS symptom management.
Results:
GWS can be difficult to differentiate from adrenal insufficiency and CS recurrence, which complicates glucocorticoid dosing and tapering regimens. Monitoring for HPA axis recovery requires both clinical and biochemical assessments. The most important intervention is reassurance to patients that GWS symptoms portend a favorable prognosis of sustained remission from CS, and GWS typically resolves as the HPA axis recovers. GWS also occurs during medical management of CS, and gradual dose titration based primarily on symptoms is essential to maintain adherence and to eventually achieve disease control. Myopathy and neurocognitive dysfunction can be chronic complications of CS that do not completely recover.
Conclusions:
Due to limited data, no guidelines have been developed for management of GWS. Nevertheless, this article provides overarching themes derived from published literature plus expert opinion and experience. Future studies are needed to better understand the pathophysiology of GWS to guide more targeted and optimal treatments.
Introduction
Endogenous neoplastic hypercortisolism - Cushing syndrome (CS) - is one of the most challenging diagnostic and management problems in clinical endocrinology. CS may be due to either a pituitary tumor (Cushing disease, CD), or a non-pituitary (ectopic) tumor secreting ACTH. ACTH-independent hypercortisolism due to unilateral or bilateral adrenal nodular disease has been increasingly recognized as an important cause of CS. Regardless of the cause of CS, the clinical manifestations are protean and include a myriad of clinical, biochemical, neurocognitive, and neuropsychiatric abnormalities. The catabolic state of hypercortisolism causes signs and symptoms including skin fragility, bruising, delayed healing, violaceous striae, muscle weakness, and low bone mass with fragility fractures. Other clinical features include weight gain, fatigue, depression, difficulty concentrating, insomnia, facial plethora, and fat redistribution to the head and neck with resultant supraclavicular and dorsocervical fullness[1]. Metabolic consequences of hypercortisolism including hypertension, diabetes, and dyslipidemia are common. In addition, women often experience hirsutism and menstrual irregularity, while men may have hypogonadism.
Management options of CS include surgery, medications, and radiation. The preferred first line treatment, regardless of source, is surgery, which offers the potential for remission[2,3,4]. The primary literature, reviews, and clinical practice guidelines for CS have traditionally focused on the diagnosis, subtyping, and surgical approach to CS. This bias derives first from the profound diagnostic challenge posed in the evaluation of cortisol production and dynamics, given that circulating cortisol follows a circadian rhythm, exhibits extensive protein binding and metabolism, and rises acutely with stress. CD and ectopic ACTH syndrome may be difficult to distinguish clinically and biochemically, and inferior petrosal sinus sampling is required in many patients to resolve this differential diagnosis. Ectopic ACTH-producing tumors can also be small, and these tumors can escape localization despite the best current methods. Although diagnosis and initial surgical remission can be achieved in the majority of patient with CS at experienced centers, up to 50% of patients with CD will require additional therapies after unsuccessful primary surgeries or recurrence up to many years later[5]. For patients who do not achieve surgical cure or who are not surgical candidates, several medical treatment options are now available. Pharmacotherapies directed at the pituitary include pasireotide[6, 7] (FDA approved) and cabergoline[8]. Adrenal steroidogenesis inhibitors such as osilodrostat[9] (FDA approved), metyrapone[10], levoketoconazole[11] (FDA approved) and ketoconazole[12], as well as the glucocorticoid antagonist, mifepristone[13] (FDA approved), are now widely used to treat CS. Pituitary radiotherapy is an additional treatment option for CD but can take months to years to lower cortisol production. Bilateral adrenalectomy (BLA) provides immediate, reliable correction of hypercortisolism but mandates life-long corticosteroid replacement therapy, and, in patients with CD, may be complicated by corticotroph tumor progression syndrome in 25–40% of patients[14].
After successful surgery for CS, the rapid onset of adrenal insufficiency (AI) is anticipated and usually portends a favorable prognosis [15,16,17,18]; however, despite the use of post-operative corticosteroid replacement, the rapid reduction in cortisol exposure often results in an enigmatic phenomenon referred to as the glucocorticoid withdrawal syndrome (GWS). This article addresses the clinical presentation and the pathogenesis of GWS, as well as its distinction from AI. When available, appropriate references are provided. Statements and guidance provided without references are derived from expert opinion and experience.
Clinical Presentation and Pathogenesis of GWS
GWS occurs following withdrawal of supraphysiologic exposure to either exogenous or endogenous glucocorticoids of at least several months duration[19]. After surgical cure of endogenous CS, GWS is usually characterized by biochemical evidence of hypothalamic-pituitary-adrenal (HPA) axis suppression with many signs and symptoms consistent with cortisol deficiency despite the use of supraphysiologic glucocorticoid replacement therapy. The degree of physical or psychologic glucocorticoid dependence experienced by patients may not correlate with the degree of HPA axis suppression[20, 21].
GWS symptom onset is typically 3–10 days postoperatively, often after the patient has been discharged from the hospital. The first symptoms of GWS vary but usually consist of myalgias, muscle weakness, fatigue, and hypersomnolence. Anorexia, nausea, and abdominal discomfort are common, but vomiting should raise concern for hyponatremia, cerebrospinal fluid leak, hydrocephalus, or other perioperative complications. Mood changes develop more gradually and range from mood swings to depression, and the fatigue with myalgias can exacerbate mood changes. An atypical depressive disorder has been described in many patients after CD surgery[22]. Weight loss should ensue in most patients but gradually and proportionate to the reduction in glucocorticoid exposure. It is important to complete a thorough symptom review and physical exam at postoperative visits, as the differentiation between GWS and bona fide AI – and even between GWS and recurrence of CS – can be challenging (Fig. 1). All three conditions are associated with symptoms of myalgias, weakness, and fatigue; however, rapid weight loss, hypoglycemia, and hypotension are suggestive of AI and the need for an increase in the glucocorticoid dose. In parallel, hypersomnia is more suggestive of GWS, while insomnia is more associated with recurrence of CS. Given the anticipation of GWS onset shortly after discharge and the potential for hyponatremia during this time, a widely employed strategy is a generous glucocorticoid dose for the first 2–3 weeks, at least until the first postoperative outpatient visit (Table 1).
Fig. 1 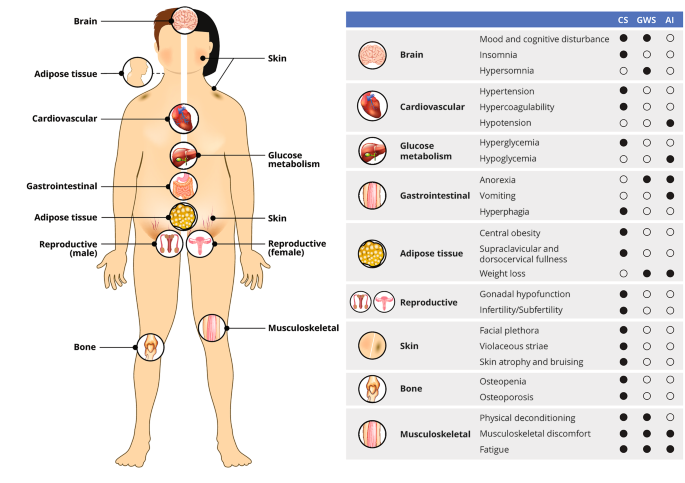
Overlapping clinical features of Cushing syndrome (CS), glucocorticoid withdrawal syndrome (GWS), and adrenal insufficiency (AI)
Table 1 Glucocorticoid Therapy Options After Surgery for CS The mechanisms responsible for the precipitation of the GWS after surgery for CS and the variability in its manifestations are not completely understood, yet alterations in the regulation of cortisol and cortisol-responsive genes appear to contribute. Down-regulation of corticotropin-releasing hormone (CRH) and proopiomelanocortin (POMC) expression, combined with up-regulation of cytokines and prostaglandins are likely to be important components of GWS. Low CRH has been associated with atypical depression[23], and CRH levels in cerebrospinal fluid of patients with CD are significantly lower compared to healthy subjects[24]. CRH suppression gradually resolves after surgical cure over 12 months during glucocorticoid replacement[25], illustrative of the slow recovery process. The expression of POMC, the ACTH precursor molecule, is also suppressed with chronic glucocorticoid exposure[26], and the normalization of POMC-associated peptides mirrors HPA axis recovery[19]. In the acute phase of glucocorticoid withdrawal, interleukins IL-6 and IL-1β, as well as tumor-necrosis factor alpha (TNFα) have been observed to rise[27], suggesting that glucocorticoid-mediated suppression of cytokines and prostaglandins is then released in GWS, and these cytokines induce the associated flu-like symptoms. Glucocorticoid replacement with dexamethasone 0.5 mg/d reduced but did not normalize IL-6 after 4–5 days[27], consistent with resistance to suppression during GWS.
Acute Care: Perioperative Planning, Coaching, and Management
For patients with CD, transsphenoidal surgery performed by an experienced surgeon achieves remission in about 80% of pituitary microadenomas and 60% of macroadenomas[28,29,30,31]. Post-operative AI and GWS are some of the most challenging phases of management for endocrinologists and one of the most disheartening for CS patients. Many patients report feeling unprepared for the postsurgical recovery process[32]. For these reasons, it is important to prepare the patient prior to surgery for the difficult months ahead, and the same considerations apply to the commencement of medical therapies, as will be discussed later. On the one hand, more potent glucocorticoids and higher doses reliably mitigate symptoms, but on the other hand, substitution of exogenous for endogenous CS delays recovery of the HPA axis and perpetuates CS-related co-morbidities. Limited data that compare management strategies preclude evidence-based decisions, yet some themes can be derived from expert opinion and extensive experience from CS centers.
In centers dedicated to the management of CS, surgeons and endocrinologists work closely together through all phases of the process. Although the goal of primary surgery for CD is adenoma resection, the tumor might not be found and/or removed completely after initial exploration. To prepare for this possibility, the surgeon should determine in advance with the patient and endocrinologist what to do next in this situation – dissect further, perform a hypophysectomy or hemi-hypophysectomy, or stop the operation. The plan for perioperative testing and glucocorticoid treatment varies widely among centers. The conundrum faced in the immediate perioperative period is that withholding glucocorticoids allows for rapid testing and demonstration of remission; however, complete resection of the causative tumor causes AI from prolonged suppression of the HPA axis and concerns for acute decompensation. Abundant evidence has shown that post-pituitary adenomectomy patients are not at risk for an adrenal crisis when monitored closely in an intensive care unit or equivalent setting[33]. Many studies have confirmed that post-operative AI almost always suggests a remission of CD[15,16,17,18, 34]. A standard protocol includes securing serum electrolytes and cortisol, plasma ACTH, capillary blood glucose, blood pressure, and urine specific gravity every 6 h for 24–48 h while withholding all glucocorticoids. Consecutive serum cortisol values less than 2–5 µg/dL (we use < 3 µg/dL) are sufficient to document successful tumor resection and to begin glucocorticoid therapy[35]. Post-operative signs and symptoms of AI including vomiting, hyponatremia, hypoglycemia, and hypotension should also mandate immediate glucocorticoid support. Although not clinically useful in the immediate post-operative period, some investigators have shown that low ACTH and DHEAS levels may be better predictors of long-term remission than serum cortisol[36]. A similar strategy for the management of possible post-operative AI/GWS following unilateral adrenalectomy for nodular adrenal disease has recently been reported. A post-operative day 1 basal cortisol and its response to cosyntropin stimulation can reliably segregate those patients with HPA axis suppression requiring cortisol replacement from those with an intact HPA axis who do not need to be discharged with glucocorticoid therapy[37].
Once remission is achieved, exogenous glucocorticoid replacement should be initiated and maintained during the months required for HPA axis recovery. Several glucocorticoids and dosing options are available (Table 1), and the initial dose is generally 3- to 4-fold higher than the physiologic range and graded based on age, comorbidities, and severity of disease. Fludrocortisone acetate should also be initiated following BLA for patients who receive glucocorticoids other than hydrocortisone, the only glucocorticoid with mineralocorticoid activity. By comparison, post-BLA patients receiving supraphysiologic hydrocortisone doses usually do not need mineralocorticoid support until their dose is tapered to near physiologic replacement. In the acute postoperative period, several medical comorbidities accompanying CS may reverse rapidly and require medication adjustments[35]. In particular, insulin and oral hypoglycemic drugs, potassium-sparing diuretics such as spironolactone, and other cardiovascular drugs are typically tapered or discontinued as glucose counter-regulation and electrolyte balance change rapidly upon cortisol reduction. Due to the high risk of postoperative venous thromboembolism[38,39,40], prophylaxis is frequently recommended and continued for several weeks after discharge. Posterior pituitary manipulation can disturb water balance and result in serum sodium alterations, including transient or permanent central diabetes insipidus, and in rare cases the triphasic response of diabetes insipidus, followed by syndrome of inappropriate secretion of antidiuretic hormone (SIADH), and finally permanent diabetes insipidus[41, 42]. In the first week or two after discharge, the most common cause for readmission is hyponatremia[43, 44], although the mechanisms responsible for this transient SIADH state are not known. For this reason, patients should be instructed to drink only when thirsty and not as an alternative to solid foods or for social reasons for 7–10 days after the surgery. Both diabetes insipidus and SIADH may not manifest for weeks after surgery; consequently, serum sodium should be monitored after hospital discharge as well [42].
Subacute Care: The GWS and HPA Axis Recovery
When managing GWS symptoms, it is important to repeatedly emphasize to the patient that not only are GWS symptoms to be expected, but in fact these manifestations portend a favorable prognosis of sustained remission from CS. The most important treatment intervention is frequent reassurance to the patient that GWS typically resolves as the HPA axis recovers. Family members must be included in the conversation to help provide as much support as possible, as patients report that support from family and friends is the most helpful coping mechanism during the recovery process[32]. When appropriate, it may be necessary to provide the patient with temporary disability documentation, since GWS symptoms may be so severe to preclude gainful employment. The patient must know that the myalgias reflect the body’s attempts to repair the muscle damage, similar to the soreness experienced the day after resistance weight training, and these aches will eventually subside. Due to the challenges of differentiating between GWS and AI, a higher glucocorticoid dose can be briefly trialed to assess if this increased glucocorticoid exposure improves symptoms, but late-day dosing should be avoided to support recovery of the circadian rhythm. In parallel, the patient should be encouraged to adequately rest, particularly going to sleep early but limiting daytime sleep to short naps.
Several other classes of medications can be trialed to target specific patient symptoms (Table 2). Antidepressants such as fluoxetine, sertraline, and trazodone might help to improve mood, sleep and appetite. A non-steroidal anti-inflammatory medication to address the musculoskeletal discomfort might be used early in the GWS, with the cyclooxygenase type 2 (COX-2) inhibitor celecoxib (100–200 mg once or twice daily) preferred when several weeks of daily treatment is needed, generally not more than 3 months. With anorexia and reduced food intake, adequate protein intake is necessary to allow muscle recovery. Egg whites, nuts, and lean meats are nutritionally dense and generally easy to tolerate despite poor appetite.
Table 2 Pharmacotherapy and Ancillary Treatment Options for GWS Symptoms Following surgical remission, the duration of glucocorticoid taper can vary from 6 to 12 months or more, depending on age, severity of disease, and duration of disease [45, 46]. Monitoring for HPA axis recovery involves both clinical and biochemical assessments. Since the HPA axis is likely to remain suppressed with prolonged supraphysiologic glucocorticoid replacement, the first goal is to shift from all-day dosing to a circadian schedule as soon as possible, such as hydrocortisone 20 mg on rising and 10 mg in the early afternoon by 2–6 weeks after surgery. The advantages of hydrocortisone include rapid absorption for symptom mitigation, the ability to measure serum cortisol as a measure of drug exposure when helpful, and the relatively short half-life [47], which ensures a glucocorticoid-free period in the early morning when it is most critical to avoid prolonged HPA axis suppression and to enhance recovery. The second goal, which should not be attempted until GWS symptoms – particularly the anorexia and myalgias – are considerably improved, is to limit replacement to a single morning dose.
Biochemical assessment should begin once patients are taking a physiologic dose of glucocorticoid replacement (total daily dose of hydrocortisone 15 to 20 mg per day) and clinically feel well enough to begin the final stage to discontinuation of glucocorticoid replacement (Fig. 2). Biochemical evaluation begins with basal testing, and dynamic assessment of adrenal function might be necessary to confirm completion of recovery. For basal testing, patients should not take their afternoon hydrocortisone dose (if prescribed) the day before testing and then have a blood draw by 0830 prior to the morning hydrocortisone dose on the day of testing. While a serum cortisol alone is adequate to taper hydrocortisone, a simultaneous plasma ACTH assists in gauging the state of HPA axis recovery. Often the ACTH and cortisol rise gradually in parallel, but sometimes the ACTH rises above the normal range despite a low cortisol, which indicates recovery of the hypothalamus (CRH neuron) and pituitary corticotrophs in advance of adrenal function. Serum DHEAS can remain suppressed for months to years after cortisol normalization, and a low DHEAS does not indicate continued HPA axis suppression. A rapid rise in DHEAS, in contrast, is concerning for disease recurrence, but a slow drift to a measurable amount in parallel with the cortisol rise is consistent with HPA axis recovery. Periodic assessment of electrolytes is prudent to screen for hyponatremia and hypo- or hyperkalemia as medications are changed, particularly diuretics. Hypercalcemia that is parathyroid-hormone independent might be observed during the recovery phase, probably related to the rise in cytokines that accompany resolution of hypercortisolemia[48, 49].
Fig. 2 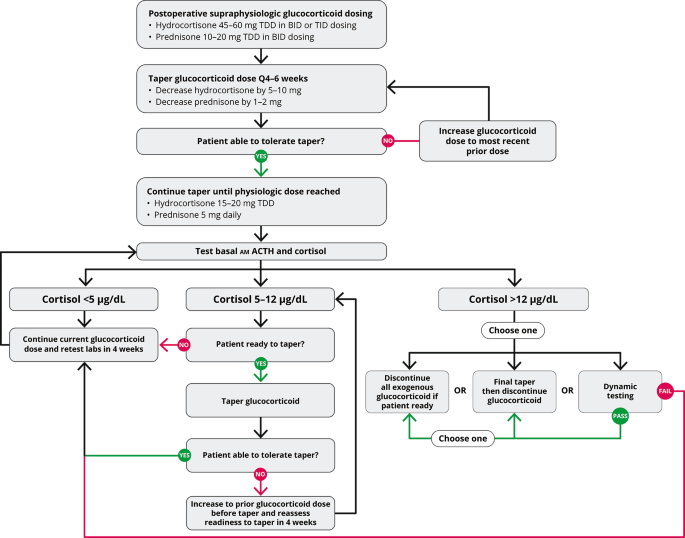
Glucocorticoid withdrawal algorithm. TDD, total daily dose
Basal testing is performed at 4- to 6-week intervals during glucocorticoid replacement. A rule of thumb is that the AM cortisol in µg/dL plus the morning dose of hydrocortisone in milligrams should sum to 15–20. Thus, once endogenous cortisol production is measurable, the hydrocortisone dose should be not more than 20 mg on arising. Once the AM cortisol rises to near 5 and then 10 µg/dL, the AM hydrocortisone dose is dropped to 15 and then 10 mg, respectively. Once the AM cortisol is 12–14 µg/dL, recovery is essentially complete, and the morning hydrocortisone dose is dropped to 5 mg for 4–6 weeks and then stopped or held for dynamic testing (Fig. 2). A clinical pearl related to HPA axis recovery is that patients who state that they are finally feeling better and getting over the GWS usually have started to make some endogenous cortisol, yet not enough to stop glucocorticoid tapering. Nevertheless, a smidgeon of endogenous cortisol production with the waning of GWS symptoms is a harbinger that HPA axis recovery is imminent. If basal testing is equivocal, dynamic testing might be necessary. The gold standard testing for central AI is the insulin tolerance test, which is rarely used, and metyrapone testing might be employed once the basal cortisol is > 10 µg/dL. Although designed to test for primary adrenal insufficiency, the cosyntropin stimulation test is often employed in this setting due to greater availability, simplicity, and safety than insulin or metyrapone testing. The duration of full HPA axis recovery can be highly variable depending on the individual and postoperative glucocorticoid dosing[50].
GWS During Medical Management of CS
Patients who are not surgical candidates or do not have successful remission of CS following surgery may be offered medical treatment or BLA. After BLA, the GWS will ensue without eventual recovery of the HPA axis, so glucocorticoids are tapered until a chronic physiologic replacement dose is reached as described previously. With medical management, patients might also experience GWS, particularly at the onset of treatment. Therefore, patients must be counseled that the typical symptoms of fatigue, myalgias, and anorexia are not only possible but indeed expected, rather than “side effects” of the medication, with two caveats. First, as described for glucocorticoid replacement following surgical remission, the endocrinologist must distinguish GWS from AI due to over-treatment of CS. The same parameters of vomiting, hypotension, and hypoglycemia favor inadequate cortisol exposure and the need for dose reduction or treatment pause and/or supplementation with a potent glucocorticoid such as dexamethasone to reverse an acute event. Second, known adverse effects of the specific drug in use should be considered and excluded. The quandary of distinguishing GWS from over-treatment raises an important principle of medical management: under-dose initially and gauge primarily the severity of GWS symptoms in the first several days. The initial goal of medical therapy is not to rapidly achieve normal cortisol milieu, but rather to “dial in” just enough inhibition of cortisol production or receptor antagonism to precipitate mild to moderate GWS symptoms. Once GWS symptoms appear and/or a typical dose of the medication is achieved, further assessments, including glucose, serum cortisol and/or UFC (except when treated with mifepristone), clinical appearance, and body weight are conducted while the dose is maintained constant until GWS symptoms begin to dissipate. If the patient is not experiencing adequate clinical and/or biochemical benefit from the medication in the absence of GWS symptoms, the dose is gradually raised incrementally. This iterative process might require periodic dose reduction or perhaps even temporarily discontinuing the medication if the patient’s daily living activities are affected at any point in the process.
For several medications, a block-and-replacement strategy is an option[3], particularly for very compliant patients for whom a priority is placed on avoidance of over-treatment. This strategy resembles thionamide-plus-levothyroxine therapy for the treatment of Graves disease. The patient is given both a generous dose of medication to completely block endogenous glucocorticoid production, plus simultaneous exogenous glucocorticoid therapy, titrated to replacement dose or greater. This approach allows for greater control over glucocorticoid exposure and low risk of AI, as long as the patient always takes both medications each day. Long-acting pasireotide, for example, would not be an appropriate drug for the block-and-replace strategy. Based on the drug mechanism of action, this block-and-replace strategy is feasible with ketoconazole or levoketoconazole, the 11β-hydroxylase inhibitors osilodrostat and metyrapone, and the adrenolytic agent mitotane (the latter three are off-label uses). Alternatively, the patient might be given a double replacement dose of glucocorticoid to take only if symptoms concerning for over-treatment occur, and the medical therapy for hypercortisolemia is then interrupted until the patient communicates with the endocrinologist.
Treatment monitoring with medical management includes biochemical and symptom assessment. For all medications other than mifepristone, normalization of 24-hour UFC is the minimal goal [2]. Basal morning cortisol and late-night salivary cortisol may be more challenging to interpret in the setting of diurnal rhythm loss characteristic of CS. Because mifepristone blocks glucocorticoid receptors, ACTH and cortisol increase with treatment for most forms of CS; dose titration therefore relies on assessment of clinical features, glycemia, body weight, and other metabolic parameters [2]. For occult tumors, periodic imaging to screen for a surgical target and/or tumor regrowth is prudent, and a pause in treatment for repeat surgery might be indicated.
The End Game: Comprehensive Recovery for the Patient with CS
Besides navigating the GWS and shepherding recovery of the HPA axis, recovery from co-morbidities of CS must be addressed to the extent possible. Hypertension, hyperglycemia, hypokalemia, and dyslipidemia often improve substantially but do not always resolve. Insomnia, skin thinning and bruising, and risk of thrombosis also generally resolve, and associated treatments might be discontinued. Although there is usually an improvement in bone density and decreased fracture risk following correction of CS, anabolic and/or anti-resorptive therapies may be warranted in some patients. The deformities of vertebral compression fractures may be permanent, and some authors have recommended the use of vertebroplasty for symptom relief[51]. Violaceous striae and chronic skin tears might heal with hyperpigmentation, leaving “the scars of Cushing’s,” which can persist for a lifetime. These milestones or minor victories can be used as evidence of healing and encouragement for the patient during the dark days of the GWS, and these changes herald further improvements. Fat redistribution and significant weight loss take some weeks to manifest and usually follow next.
The myopathy from CS is an example of a co-morbidity that rarely improves without targeted treatment, and the German Cushing’s Registry has provided evidence for chronic muscle dysfunction following cure of CS[52]. Recent data indicate that a low IGF-1 after curative surgery is associated with long-term myopathy [53]. This persistent myopathy is a common source of chronic fatigue following HPA axis recovery, which is unresponsive to glucocorticoids. For these reasons, an important ancillary modality is physical therapy, and an ideal time to initiate this treatment is at the first signs of HPA axis recovery when the GWS symptoms have subsided. A complete evaluation from an experienced physical therapist should focus on core and proximal muscle strength, balance, and other factors that limit function. Exercises targeting these factors (stand on one foot, sit-to-stand, straight-arm raises with 1- to 5-pound weights) rather than traditional gym exercises (arm curls, bench press, treadmill) are necessary to restore functional status and avoid frustration and injury when the patient is not yet prepared for the latter stages of recovery. Professional supervision of this initial phase is a critical component of the recovery process, and failure to attend to musculoskeletal rehabilitation – as would be routine following survival of a critical illness – risks long-term morbidities from a curable disease.
Patients with CS often complain of cognitive defects, which usually improve but may not completely recover following treatment[54, 55]. Glucocorticoids are toxic to the hippocampus, and both rats treated with high-dose corticosterone and patients with CD experience reductions in hippocampal volume, which does not completely return to normal even with correction of hypercortisolemia[56, 57]. Because the hippocampus is an important brain region for memory, the main complaint is impaired formation of new memories and recall of recent events. When significant cognitive dysfunction persists, a formal neuropsychologic testing session is prudent, both to screen for additional sources of memory loss (degenerative brain diseases) and to identify aspects that might be amenable to functional management approaches. Cognitive therapy can be effective for mental health and overall disease coping strategies as well.
Finally, for patients undergoing transsphenoidal surgery for CD, complications associated with pituitary surgeries in general should also be considered. Anterior pituitary hormone axes should be assessed biochemically and symptomatically for hypothyroidism and hypogonadism, as hypopituitarism is an independent predictor of decreased quality of life after surgical cure [58]. Hypopituitarism can not only complicate the assessment of GWS with overlapping symptoms such as fatigue, but treatment of hypopituitarism can also be important for GWS recovery. Prior to initiating physical therapy, testosterone replacement in male patients with hypogonadism should be optimized. Hypothyroidism can contribute to hyponatremia and can also slow the metabolism of glucocorticoids. Therefore, optimizing the treatment of hypothyroidism and hypogonadism prior to completing glucocorticoid taper is prudent. Growth hormone deficiency may also be evaluated in symptomatic patients in the setting of other anterior pituitary hormone deficiencies, although formal evaluation is best delayed for at least 6–12 months when HPA axis recovery has occurred or at least the glucocorticoid dose is reduced to a physiologic range [2].
Summary and Final Thoughts
After a diagnosis of CS has been well established, a multidisciplinary team of endocrinologists and surgeons must design the best treatment strategy for the patient. Expectations and possible adverse side effects of surgery or pharmacotherapy should be reviewed with the patient. The GWS is a very difficult concept for patients to understand. It seems inconceivable to them that they could possibly feel worse (and that this is a good omen) six weeks after resolution of their hypercortisolism than they do pre-operatively; however, there are no studies that address whether comprehensive pre-operative patient education regarding GWS has any impact on the patient’s post-operative perception and outcome after successful surgery. An addiction metaphor is sometimes helpful: the patient’s body and brain has become addicted to steroids (cortisol) and after steroids are abruptly reduced, their body and brain are dysphoric — much like removal of any other addictive substance (e.g., opioids, alcohol, nicotine). The patient and their care team need to know that this treatment odyssey will be a marathon, not a sprint. It may take as long as 12–18 months for patients to have full HPA axis recovery, regression of GWS, and, most importantly, resolution of the devastating effects of chronic excessive glucocorticoid exposure.
Conclusions
GWS following surgery or during medical treatment of CS can be challenging to manage. There are currently no standard guidelines for management of GWS, but various available medical and ancillary therapies are discussed here. Studies are needed to better understand the pathophysiology of GWS to guide more targeted treatments. There may be yet unrecognized steroids produced by the adrenal glands, the withdrawal of which contributes to GWS symptoms[59]. Future observational and interventional studies would be beneficial for identifying optimal management options.
References
-
Carroll TB, Findling JW (2010) The diagnosis of Cushing’s syndrome. Rev Endocr Metab Disord 11:147–153. https://doi.org/10.1007/s11154-010-9143-3
-
Fleseriu M, Auchus R, Bancos I et al (2021) Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol 9:847–875. https://doi.org/10.1016/S2213-8587(21)00235-7
-
Nieman LK, Biller BMK, Findling JW et al (2015) Treatment of Cushing’s Syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 100:2807–2831. https://doi.org/10.1210/jc.2015-1818
-
Biller BMK, Grossman AB, Stewart PM et al (2008) Treatment of adrenocorticotropin-dependent Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab 93:2454–2462. https://doi.org/10.1210/jc.2007-2734
-
Geer EB, Shafiq I, Gordon MB et al (2017) BIOCHEMICAL CONTROL DURING LONG-TERM FOLLOW-UP OF 230 ADULT PATIENTS WITH CUSHING DISEASE: A MULTICENTER RETROSPECTIVE STUDY. Endocr Pract 23:962–970. https://doi.org/10.4158/EP171787.OR
-
Colao A, Petersenn S, Newell-Price J et al (2012) A 12-Month Phase 3 Study of Pasireotide in Cushing’s Disease. N Engl J Med 366:914–924. https://doi.org/10.1056/NEJMoa1105743
-
Lacroix A, Gu F, Gallardo W et al (2018) Efficacy and safety of once-monthly pasireotide in Cushing’s disease: a 12 month clinical trial. Lancet Diabetes Endocrinol 6:17–26. https://doi.org/10.1016/S2213-8587(17)30326-1
-
Pivonello R, De Martino MC, Cappabianca P et al (2009) The medical treatment of Cushing’s disease: effectiveness of chronic treatment with the dopamine agonist cabergoline in patients unsuccessfully treated by surgery. J Clin Endocrinol Metab 94:223–230. https://doi.org/10.1210/jc.2008-1533
-
Pivonello R, Fleseriu M, Newell-Price J et al (2020) Efficacy and safety of osilodrostat in patients with Cushing’s disease (LINC 3): a multicentre phase III study with a double-blind, randomised withdrawal phase. Lancet Diabetes Endocrinol 8:748–761. https://doi.org/10.1016/S2213-8587(20)30240-0
-
Ceccato F, Zilio M, Barbot M et al (2018) Metyrapone treatment in Cushing’s syndrome: a real-life study. Endocrine 62:701–711. https://doi.org/10.1007/s12020-018-1675-4
-
Fleseriu M, Pivonello R, Elenkova A et al (2019) Efficacy and safety of levoketoconazole in the treatment of endogenous Cushing’s syndrome (SONICS): a phase 3, multicentre, open-label, single-arm trial. Lancet Diabetes Endocrinol 7:855–865. https://doi.org/10.1016/S2213-8587(19)30313-4
-
Castinetti F, Guignat L, Giraud P et al (2014) Ketoconazole in Cushing’s disease: is it worth a try? J Clin Endocrinol Metab 99:1623–1630. https://doi.org/10.1210/jc.2013-3628
-
Fleseriu M, Biller BMK, Findling JW et al (2012) Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing’s syndrome. J Clin Endocrinol Metab 97:2039–2049. https://doi.org/10.1210/jc.2011-3350
-
Reincke M, Albani A, Assie G et al (2021) Corticotroph tumor progression after bilateral adrenalectomy (Nelson’s syndrome): systematic review and expert consensus recommendations. Eur J Endocrinol 184:P1–P16. https://doi.org/10.1530/EJE-20-1088
-
Lindsay JR, Oldfield EH, Stratakis CA, Nieman LK (2011) The Postoperative Basal Cortisol and CRH Tests for Prediction of Long-Term Remission from Cushing’s Disease after Transsphenoidal Surgery. J Clin Endocrinol Metab 96:2057–2064. https://doi.org/10.1210/jc.2011-0456
-
Hameed N, Yedinak CG, Brzana J et al (2013) Remission rate after transsphenoidal surgery in patients with pathologically confirmed Cushing’s disease, the role of cortisol, ACTH assessment and immediate reoperation: a large single center experience. Pituitary 16:452–458. https://doi.org/10.1007/s11102-012-0455-z
-
Ramm-Pettersen J, Halvorsen H, Evang JA et al (2015) Low immediate postoperative serum-cortisol nadir predicts the short-term, but not long-term, remission after pituitary surgery for Cushing’s disease. BMC Endocr Disord 15:62. https://doi.org/10.1186/s12902-015-0055-9
-
Ironside N, Chatain G, Asuzu D et al (2018) Earlier post-operative hypocortisolemia may predict durable remission from Cushing’s disease. Eur J Endocrinol 178:255–263. https://doi.org/10.1530/EJE-17-0873
-
Hochberg Z, Pacak K, Chrousos GP (2003) Endocrine Withdrawal Syndromes. Endocr Rev 24:523–538. https://doi.org/10.1210/er.2001-0014
-
Dixon RB, Christy NP (1980) On the various forms of corticosteroid withdrawal syndrome. Am J Med 68:224–230. https://doi.org/10.1016/0002-9343(80)90358-7
-
AMATRUDA TT ND JR (1965) Certain Endocrine and Metabolic Facets of the Steroid Withdrawal Syndrome. J Clin Endocrinol Metab 25:1207–1217. https://doi.org/10.1210/jcem-25-9-1207
-
Dorn LD, Burgess ES, Friedman TC et al (1997) The Longitudinal Course of Psychopathology in Cushing’s Syndrome after Correction of Hypercortisolism. J Clin Endocrinol Metab 82:912–919. https://doi.org/10.1210/jcem.82.3.3834
-
Chrousos GP, Gold PW (1992) The Concepts of Stress and Stress System Disorders: Overview of Physical and Behavioral Homeostasis. JAMA 267:1244–1252. https://doi.org/10.1001/jama.1992.03480090092034
-
Kling MA, Roy A, Doran AR et al (1991) Cerebrospinal fluid immunoreactive corticotropin-releasing hormone and adrenocorticotropin secretion in Cushing’s disease and major depression: potential clinical implications. J Clin Endocrinol Metab 72:260–271. https://doi.org/10.1210/jcem-72-2-260
-
Gomez MT, Magiakou MA, Mastorakos G, Chrousos GP (1993) The pituitary corticotroph is not the rate limiting step in the postoperative recovery of the hypothalamic-pituitary-adrenal axis in patients with Cushing syndrome. J Clin Endocrinol Metab 77:173–177. https://doi.org/10.1210/jcem.77.1.8392083
-
Young EA, Kwak SP, Kottak J (1995) Negative feedback regulation following administration of chronic exogenous corticosterone. J Neuroendocrinol 7:37–45. https://doi.org/10.1111/j.1365-2826.1995.tb00665.x
-
Papanicolaou DA, Tsigos C, Oldfield EH, Chrousos GP (1996) Acute glucocorticoid deficiency is associated with plasma elevations of interleukin-6: does the latter participate in the symptomatology of the steroid withdrawal syndrome and adrenal insufficiency? J Clin Endocrinol Metab 81:2303–2306. https://doi.org/10.1210/jcem.81.6.8964868
-
Ciric I, Zhao J-C, Du H et al (2012) Transsphenoidal surgery for Cushing disease: experience with 136 patients. Neurosurgery 70:70–80 discussion 80–81. https://doi.org/10.1227/NEU.0b013e31822dda2c
-
Alexandraki KI, Kaltsas GA, Isidori AM et al (2013) Long-term remission and recurrence rates in Cushing’s disease: predictive factors in a single-centre study. Eur J Endocrinol 168:639–648. https://doi.org/10.1530/EJE-12-0921
-
Capatina C, Hinojosa-Amaya JM, Poiana C, Fleseriu M (2020) Management of patients with persistent or recurrent Cushing’s disease after initial pituitary surgery. Expert Rev Endocrinol Metab 15:321–339. https://doi.org/10.1080/17446651.2020.1802243
-
Stroud A, Dhaliwal P, Alvarado R et al (2020) Outcomes of pituitary surgery for Cushing’s disease: a systematic review and meta-analysis. Pituitary 23:595–609. https://doi.org/10.1007/s11102-020-01066-8
-
Acree R, Miller CM, Abel BS et al (2021) Patient and Provider Perspectives on Postsurgical Recovery of Cushing Syndrome. J Endocr Soc 5:bvab109. https://doi.org/10.1210/jendso/bvab109
-
AbdelMannan D, Selman WR, Arafah BM (2010) Peri-operative management of Cushing’s disease. Rev Endocr Metab Disord 11:127–134. https://doi.org/10.1007/s11154-010-9140-6
-
Costenaro F, Rodrigues TC, Rollin GAF et al (2014) Evaluation of Cushing’s disease remission after transsphenoidal surgery based on early serum cortisol dynamics. Clin Endocrinol (Oxf) 80:411–418. https://doi.org/10.1111/cen.12300
-
Varlamov EV, Vila G, Fleseriu M (2022) Perioperative Management of a Patient With Cushing Disease. J Endocr Soc 6:bvac010. https://doi.org/10.1210/jendso/bvac010
-
El Asmar N, Rajpal A, Selman WR, Arafah BM (2018) The Value of Perioperative Levels of ACTH, DHEA, and DHEA-S and Tumor Size in Predicting Recurrence of Cushing Disease. J Clin Endocrinol Metab 103:477–485. https://doi.org/10.1210/jc.2017-01797
-
DeLozier OM, Dream SY, Findling JW et al (2022) Selective Glucocorticoid Replacement Following Unilateral Adrenalectomy for Hypercortisolism and Primary Aldosteronism. J Clin Endocrinol Metab 107:e538–e547. https://doi.org/10.1210/clinem/dgab698
-
Stuijver DJF, van Zaane B, Feelders RA et al (2011) Incidence of venous thromboembolism in patients with Cushing’s syndrome: a multicenter cohort study. J Clin Endocrinol Metab 96:3525–3532. https://doi.org/10.1210/jc.2011-1661
-
van der Pas R, Leebeek FWG, Hofland LJ et al (2013) Hypercoagulability in Cushing’s syndrome: prevalence, pathogenesis and treatment. Clin Endocrinol (Oxf) 78:481–488. https://doi.org/10.1111/cen.12094
-
van der Pas R, de Bruin C, Leebeek FWG et al (2012) The hypercoagulable state in Cushing’s disease is associated with increased levels of procoagulant factors and impaired fibrinolysis, but is not reversible after short-term biochemical remission induced by medical therapy. J Clin Endocrinol Metab 97:1303–1310. https://doi.org/10.1210/jc.2011-2753
-
Kristof RA, Rother M, Neuloh G, Klingmüller D (2009) Incidence, clinical manifestations, and course of water and electrolyte metabolism disturbances following transsphenoidal pituitary adenoma surgery: a prospective observational study: Clinical article. J Neurosurg 111:555–562. https://doi.org/10.3171/2008.9.JNS08191
-
Yuen KCJ, Ajmal A, Correa R, Little AS (2019) Sodium Perturbations After Pituitary Surgery. Neurosurg Clin 30:515–524. https://doi.org/10.1016/j.nec.2019.05.011
-
Ghiam MK, Chyou DE, Dable CL et al (2021) 30-Day Readmissions and Coordination of Care Following Endoscopic Transsphenoidal Pituitary Surgery: Experience with 409 Patients. J Neurol Surg Part B Skull Base. https://doi.org/10.1055/s-0041-1729980
-
Bohl MA, Ahmad S, Jahnke H et al (2016) Delayed Hyponatremia Is the Most Common Cause of 30-Day Unplanned Readmission After Transsphenoidal Surgery for Pituitary Tumors. Neurosurgery 78:84–90. https://doi.org/10.1227/NEU.0000000000001003
-
Doherty GM, Nieman LK, Cutler GB et al (1990) Time to recovery of the hypothalamic-pituitary-adrenal axis after curative resection of adrenal tumors in patients with Cushing’s syndrome. Surgery 108:1085–1090
-
Sippel RS, Elaraj DM, Kebebew E et al (2008) Waiting for change: Symptom resolution after adrenalectomy for Cushing’s syndrome. Surgery 144:1054–1061. https://doi.org/10.1016/j.surg.2008.08.024
-
Derendorf H, Möllmann H, Barth J et al (1991) Pharmacokinetics and Oral Bioavailability of Hydrocortisone. J Clin Pharmacol 31:473–476. https://doi.org/10.1002/j.1552-4604.1991.tb01906.x
-
Suzuki K, Nonaka K, Ichihara K et al (1986) Hypercalcemia in Glucocorticoid Withdrawal. Endocrinol Jpn 33:203–209. https://doi.org/10.1507/endocrj1954.33.203
-
Oyama Y, Iwafuchi Y, Narita I (2021) A case of hypercalcemia because of adrenal insufficiency induced by glucocorticoid withdrawal in a patient undergoing hemodialysis. CEN Case Rep. https://doi.org/10.1007/s13730-021-00619-5
-
Berr CM, Di Dalmazi G, Osswald A et al (2015) Time to Recovery of Adrenal Function After Curative Surgery for Cushing’s Syndrome Depends on Etiology. J Clin Endocrinol Metab 100:1300–1308. https://doi.org/10.1210/jc.2014-3632
-
Gad HEM, Ismail AM (2020) The role of vertebroplasty in steroid-induced vertebral osteoporotic fractures. Egypt Spine J 35:41–52. https://doi.org/10.21608/esj.2020.34844.1140
-
Vogel F, Braun LT, Rubinstein G et al (2020) Persisting Muscle Dysfunction in Cushing’s Syndrome Despite Biochemical Remission. J Clin Endocrinol Metab 105:e4490–e4498. https://doi.org/10.1210/clinem/dgaa625
-
Vogel F, Braun L, Rubinstein G et al (2021) Patients with low IGF-I after curative surgery for Cushing’s syndrome have an adverse long-term outcome of hypercortisolism-induced myopathy. Eur J Endocrinol 184:813–821. https://doi.org/10.1530/EJE-20-1285
-
Andela CD, van Haalen FM, Ragnarsson O et al (2015) MECHANISMS IN ENDOCRINOLOGY: Cushing’s syndrome causes irreversible effects on the human brain: a systematic review of structural and functional magnetic resonance imaging studies. Eur J Endocrinol 173:R1–R14. https://doi.org/10.1530/EJE-14-1101
-
Bride MM, Crespo I, Webb SM, Valassi E (2021) Quality of life in Cushing’s syndrome. Best Pract Res Clin Endocrinol Metab 35:101505. https://doi.org/10.1016/j.beem.2021.101505
-
Starkman MN, Gebarski SS, Berent S, Schteingart DE (1992) Hippocampal formation volume, memory dysfunction, and cortisol levels in patients with Cushing’s syndrome. Biol Psychiatry 32:756–765. https://doi.org/10.1016/0006-3223(92)90079-F
-
McEwen BS, Gould EA, Sakai RR (1992) The Vulnerability of the Hippocampus to Protective and Destructive Effects of Glucocorticoids in Relation to Stress. Br J Psychiatry 160:18–23. https://doi.org/10.1192/S0007125000296645
-
van Aken MO, Pereira AM, Biermasz NR et al (2005) Quality of Life in Patients after Long-Term Biochemical Cure of Cushing’s Disease. J Clin Endocrinol Metab 90:3279–3286. https://doi.org/10.1210/jc.2004-1375
-
Zorumski CF, Paul SM, Izumi Y et al (2013) Neurosteroids, stress and depression: potential therapeutic opportunities. Neurosci Biobehav Rev 37:109–122. https://doi.org/10.1016/j.neubiorev.2012.10.005
Acknowledgements
We thank Recordati Rare Diseases for their support with literature review and figure preparation to the authors’ designs.
Funding
XH is supported by grant T32DK07245 from the National Institutes of Diabetes and Digestive and Kidney Diseases.
Ethics declarations
Financial Interests
Dr. Auchus has received research support from Novartis Pharmaceuticals, Corcept Therapeutics, Spruce Biosciences, and Neurocrine Biosciences and has served as a consultant for Corcept Therapeutics, Janssen Pharmaceuticals, Novartis Pharmaceuticals, Quest Diagnostics, Adrenas Therapeutics, Crinetics Pharmaceuticals, PhaseBio Pharmaceuticals, OMass Therapeutics, Recordati Rare Diseases, Strongbridge Biopharma, and H Lundbeck A/S. Dr. Findling has received research support from Novartis Pharmaceuticals and has served as a consultant for Corcept Therapeutics and Recordati Rare Diseases.
Human Subjects and Animals
No human subjects or animals were used to collect data for this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Cite this article
He, X., Findling, J.W. & Auchus, R.J. Glucocorticoid Withdrawal Syndrome following treatment of endogenous Cushing Syndrome. Pituitary (2022). https://doi.org/10.1007/s11102-022-01218-y
-
Accepted
-
Published
From https://link.springer.com/article/10.1007/s11102-022-01218-y
-
 1
1
-
-
Compared with placebo, levoketoconazole improved cortisol control and serum cholesterol levels for adults with endogenous Cushing’s syndrome, according to results from the LOGICS study presented here.
Safety and efficacy of levoketoconazole (Recorlev, Xeris Biopharma) for treatment of Cushing’s syndrome were established in the pivotal phase 3, open-label SONICS study. The phase 3, double-blind LOGICS study sought to demonstrate the drug specificity of levoketoconazole in normalizing mean urinary free cortisol (mUFC) level.
“Treatment with levoketoconazole benefited patients with Cushing’s syndrome of different etiologies and a wide range in UFC elevations at baseline by frequent normalization of UFC,” Ilan Shimon, MD, professor at the Sackler Faculty of Medicine at Tel Aviv University and associate dean of the Faculty of Medicine at Rabin Medical Center and director of the Institute of Endocrinology in Israel, told Healio. “This is a valuable Cushing’s study as it includes a placebo-controlled randomized withdrawal phase.”
LOGICS participants were drawn from a cohort of 79 adults with Cushing’s syndrome with a baseline mUFC at least 1.5 times the upper limit of normal who participated in a single-arm, open-label titration and maintenance phase of approximately 14 to 19 weeks. Researchers randomly assigned 39 of those participants plus five from SONICS who had normalized mUFC levels on stable doses of levoketoconazole for at least 4 weeks to continue to receive the medication (n = 22) or to receive placebo with withdrawal of the medication (n = 22) for 8 weeks. At the end of the withdrawal period, all participants received levoketoconazole for 8 more weeks. Primary endpoint was proportion of participants who lost mUFC normalization during the randomized withdrawal period, and secondary endpoints included proportion with normalized mUFC and changes in total and LDL cholesterol at the end of the restoration period.
During the withdrawal period, 95.5% of participants receiving placebo vs. 40.9% of those receiving levoketoconazole experienced loss of mUFC response, for a treatment difference of –54.5% (95% CI, –75.7 to –27.4; P = .0002). At the end of the withdrawal period, 4.5% of participants receiving placebo vs. 50% of those receiving levoketoconazole maintained normalized mUFC, for a treatment difference of 45.5% (95% CI, 19.2-67.9; P = .0015).
Among participants who had received placebo and lost mUFC response, 60% regained normalized mUFC at the end of the restoration period.
During the withdrawal period, participants in the placebo group had increases of 0.9 mmol/L in total cholesterol and 0.6 mmol/L in LDL cholesterol vs. decreases of 0.04 mmol/L (P = .0004) and 0.006 mmol/L (P = .0056), respectively, for the levoketoconazole group. The increases seen in the placebo group were reversed when participants restarted the medication.
The most common adverse events with levoketoconazole were nausea (29%) and hypokalemia (26%). Prespecified adverse events of special interest were liver-related (10.7%), QT interval prolongation (10.7%) and adrenal insufficiency (9.5%).
“This study has led to the FDA decision to approve levoketoconazole for the treatment of Cushing’s syndrome after surgical failure or if surgery is not possible,” Shimon said.
-
1
-
-
A worldwide, observational study of adults and adolescents with growth hormone deficiency (GHD) found long-term GH replacement was safe. These findings were published in the Journal of Clinical Endocrinology & Metabolism.
Data for this long-term follow-up study were sourced from the KIMS Pfizer International Metabolic Database cohort. Patients (N=15,809) with confirmed GHD were prescribed GH by their primary care physician. Adverse events were evaluated at up to 18 years (mean, 5.3 years).
The median age of study participants was 44.8 (range, 5.6-91.2) years, 50.5% were girls or women, 94.4% were White, 57.6% were true-naive to treatment at baseline, 59.7% had pituitary or hypothalamic tumor, 21.6% had idiopathic or congenital GHD, and 67.8% had at least 2 pituitary deficiencies.
Patients were administered a mean GH dosage of 0.30±0.30 mg/d.
At year 15, patients (n=593) had a 1.7-kg/m2 increase in body mass index (BMI), a 4.3-kg increase in weight, a 0.4-cm decrease in height, a 6.2-cm increase in waist circumference, a 0.03 increase in waist to hip ratio, a 6.3-mm Hg increase in systolic blood pressure, a 1.0-mm Hg increase in diastolic blood pressure, and a 0.5-bpm decrease in heart rate.
Approximately one-half of the patients (51.2%) experienced at least 1 adverse event, but few patients (18.8%) reported treatment-related adverse events.
The most common all-cause adverse events included arthralgia (4.6%), peripheral edema (3.9%), headache (3.6%), influenza (2.8%), depression (2.8%), and recurrence of pituitary tumor (2.7%). The most common treatment-related adverse events were peripheral edema (3.1%) and arthralgia (2.6%).
The rate of all-cause (P =.0141) and related (P =.0313) adverse events was significantly related with age at enrollment, with older patients (aged ³45 years) having higher rates than younger patients.
The rate of all-cause and related adverse events was higher among patients with pituitary or hypothalamic tumor, adult-onset GHD, and insulin-like growth factor 1 standard deviation score greater than 0; those who had prior pituitary radiation treatment; and those who took a GH dosage of no more than 0.30 mg/d (all P £..014).
A total of 1934 patients discontinued treatment, and 869 patients reduced their dose due to adverse events. Study discontinuation was highest among patients with idiopathic or congenital GHD (45.0%).
At least 1 serious adverse event occurred among 4.3% of patients. The most common serious events included recurrence of pituitary tumor (n=154; 1.0%) and death (n=21; 0.1%). The highest mortality rate was observed among patients who enrolled at 45 years of age and older (4.7%).
In total, 418 patients who had no history of cancer at baseline were diagnosed with cancer after starting GH treatment, which equated to a standardized incidence ratio of 0.92 (95% CI, 0.83-1.01).
This study was limited as data were collected during routine clinical practice and no predefined windows or reporting were set.
This study found that GH replacement therapy was safe at up to an 18-year follow-up among adolescents and adults.
Disclosure: Multiple authors declared affiliations with industry. Please refer to the original article for a full list of disclosures.
Reference
Johannsson G, Touraine P, Feldt-Rasmussen U, et al. Long-term safety of growth hormone in adults with growth hormone deficiency: overview of 15,809 GH-treated patients. J Clin Endocrinol Metab. Published online April 3, 2022. doi:10.1210/clinem/dgac199
-
1
-
-
Abstract
ContextArginine-vasopressin and CRH act synergistically to stimulate secretion of ACTH. There is evidence that glucocorticoids act via negative feedback to suppress arginine-vasopressin secretion.
ObjectiveOur hypothesis was that a postoperative increase in plasma copeptin may serve as a marker of remission of Cushing disease (CD).
DesignPlasma copeptin was obtained in patients with CD before and daily on postoperative days 1 through 8 after transsphenoidal surgery. Peak postoperative copeptin levels and Δcopeptin values were compared among those in remission vs no remission.
ResultsForty-four patients (64% female, aged 7-55 years) were included, and 19 developed neither diabetes insipidus (DI) or syndrome of inappropriate anti-diuresis (SIADH). Thirty-three had follow-up at least 3 months postoperatively. There was no difference in peak postoperative copeptin in remission (6.1 pmol/L [4.3-12.1]) vs no remission (7.3 pmol/L [5.4-8.4], P = 0.88). Excluding those who developed DI or SIADH, there was no difference in peak postoperative copeptin in remission (10.2 pmol/L [6.9-21.0]) vs no remission (5.4 pmol/L [4.6-7.3], P = 0.20). However, a higher peak postoperative copeptin level was found in those in remission (14.6 pmol/L [±10.9] vs 5.8 (±1.4), P = 0.03]) with parametric testing. There was no difference in the Δcopeptin by remission status.
ConclusionsA difference in peak postoperative plasma copeptin as an early marker to predict remission of CD was not consistently present, although the data point to the need for a larger sample size to further evaluate this. However, the utility of this test may be limited to those who develop neither DI nor SIADH postoperatively.
Arginine vasopressin (AVP) and CRH act synergistically as the primary stimuli for secretion of ACTH, leading to release of cortisol [1, 2]. The role of AVP in the hypothalamic-pituitary-adrenal (HPA) axis is via release from the parvocellular neurons of the paraventricular nuclei (and possibly also from the magnocellular neurons of the paraventricular and supraoptic nuclei), the secretion of which is stimulated by stress [3-6]. AVP release results in both independent stimulation of ACTH release and potentiation of the effects of CRH [3, 7-9]. Additionally, there is evidence that glucocorticoids act by way of negative feedback to suppress AVP secretion [10, 11-20]. Further, parvocellular neurons of the hypothalamic paraventricular nuclei have been shown to increase AVP production and neurosecretory granule size after adrenalectomy, and inappropriately elevated plasma AVP has been reported in the setting of adrenal insufficiency with normalization of plasma AVP after glucocorticoid administration [21-24]. This relationship of AVP and its effect on the HPA axis has been used in the diagnostic evaluation of Cushing syndrome (CS) [14] and evaluation of remission after transsphenoidal surgery (TSS) in Cushing disease (CD) by administration of desmopressin [25].
Copeptin makes up the C-terminal portion of the AVP precursor pre-pro-AVP. Copeptin is released from the posterior pituitary in stoichiometric amounts with AVP, and because of its longer half-life in circulation, it is a stable surrogate marker of AVP secretion [26-28]. Plasma copeptin has been studied in various conditions of the anterior pituitary. In a study by Lewandowski et al, plasma copeptin was measured after administration of CRH in assessment of HPA-axis function in patients with a variety of pituitary diseases. An increase in plasma copeptin was observed only in healthy subjects but not in those with pituitary disease who had an appropriately stimulated serum cortisol, and the authors concluded that copeptin may be a sensitive marker to reveal subtle alterations in the regulation of pituitary function [7]. Although in this study and others, plasma copeptin was assessed after pituitary surgery, it has not, to the best of our knowledge, been studied as a marker of remission of CD before and after pituitary surgery [7, 29].
In this study, plasma copeptin levels were assessed as a surrogate of AVP secretion before and after TSS for treatment of CD. Because there is evidence that glucocorticoids exert negative feedback on AVP, we hypothesized that there would be a greater postoperative increase in plasma copeptin in those with CD in remission after TSS resulting from resolution of hypercortisolemia and resultant hypocortisolemia compared with those not in remission with persistent hypercortisolemia and continued negative feedback. In other words, we hypothesized that an increase in copeptin could be an early marker of remission of CD after TSS. We aimed to complete this assessment by comparison of the peak postoperative copeptin and change in copeptin from preoperative to peak postoperative copeptin for those in remission vs not in remission postoperatively.
Subjects and Methods
Subjects
Adult and pediatric patients with CD who presented at the Eunice Kennedy Shriver National Institute of Child Health and Human Development under protocol 97-CH-0076 and underwent TSS between March 2016 and July 2019 were included in the study. Exclusion criteria included a prior TSS within 6 weeks of the preoperative plasma copeptin sample or a preoperative diagnosis of diabetes insipidus, renal disease, or cardiac failure. Written informed consent was provided by patients aged 18 years and older and by legal guardians for patients aged < 18 years to participate in this study. Written informed assent was provided by patients aged 7 years to < 18 years. The 97-CH-0076 study (Investigation of Pituitary Tumors and Related Hypothalamic Disorders) has been approved by the Eunice Kennedy Shriver National Institute of Child Health and Human Development institutional review board.
Clinical and Biochemical Data
Clinical data were extracted from electronic medical records. Age, sex, body weight, body mass index (BMI), pubertal stage (in pediatric patients only), and history of prior TSS were obtained preoperatively during the admission for TSS. Clinical data obtained postoperatively included TSS date, histology, development of central diabetes insipidus (DI) or (SIADH), time from TSS to most recent follow-up, and clinical remission status at postoperative follow-up.
Preoperatively, serum sodium, 24-hour urinary free cortisol (UFC), UFC times the upper limit of normal (UFC × ULN), midnight (MN) serum cortisol, MN plasma ACTH, and 8 AM plasma ACTH were collected. Postoperatively, serum sodium, serum and urine osmolality, urine specific gravity, serum cortisol, and plasma ACTH were collected. For serum cortisol values < 1 mcg/dL, a value of 0.5 mcg/dL was assigned for the analyses; for plasma ACTH levels < 5 pg/mL, a value of 2.5 pg/mL was assigned.
Additionally, plasma copeptin levels were obtained preoperatively and on postoperative days (PODs) 1 through 8 after TSS at 8:00 AM. Peak postoperative copeptin was the highest plasma copeptin on PODs 1 through 8. The delta copeptin (Δcopeptin) was determined by subtracting the preoperative copeptin from the peak postoperative copeptin; hence, a positive change indicated a postoperative increase in plasma copeptin. Plasma copeptin was measured using an automated immunofluorescent sandwich assay on the BRAHMS Kryptor Compact PLUS Copeptin-proAVP. The limit of detection for the assay was 1.58 pmol/L, 5.7% intra-assay coefficient of variation, and 11.2% inter-assay coefficient of variation, with a lower limit of analytical measurement of 2.8 pmol/L. For those with multiple preoperative plasma copeptin values within days before surgery, an average of preoperative copeptin levels was used for analyses.
Diagnosis of CD was based on guidelines published by the Endocrine Society and as previously described for the adult and pediatric populations [30, 31]; diagnosis was further confirmed by either histologic identification of an ACTH-secreting pituitary adenoma in the resected tumor specimen, decrease in cortisol and ACTH levels postoperatively, and/or clinical remission after TSS at follow-up evaluation. All patients were treated with TSS at the National Institutes of Health Clinical Center by the same neurosurgeon. Remission after surgical therapy was based on serum cortisol of < 5 μg/dL during the immediate postoperative period, improvement of clinical signs and symptoms of cortisol excess at postoperative follow up, nonelevated 24-hour UFC at postoperative follow-up, nonelevated midnight serum cortisol at postoperative follow up when available, and continued requirement for glucocorticoid replacement at 3 to 6 months’ postoperative follow-up.
Diagnosis of SIADH was based on development of hyponatremia (serum sodium < 135 mmol/L) and oliguria (urine output < 0.5 mL/kg/h). Diagnosis of DI was determined by development of hypernatremia (serum sodium > 145 mmol/L), dilute polyuria (urine output > 4 mL/kg/h), elevated serum osmolality, and low urine osmolality.
Statistical Analyses
Results are presented as median (interquartile range [IQR], calculated as 25th percentile-75th percentile) or mean ± SD, as appropriate, and frequency (percentage). Where appropriate, we compared results using parametric or nonparametric testing; however, the median (IQR) and the mean ± SD were both reported to allow for comparisons with the appropriate testing noted. Subgroup analyses were completed comparing those who developed water balance disorders included patients who developed DI only (but not SIADH), those who developed SIADH only (but not DI), and those with no water balance disorder; hence, for these subgroup analyses, those who developed both DI and SIADH postoperatively (n = 4) were excluded. Preoperative copeptin, peak postoperative copeptin, and Δcopeptin were compared between those with and without remission at follow-up, using either t test or Wilcoxon rank-sum test, depending on the distribution of data. These were done in all patients combined, as well as within each subgroup. The same tests were used for comparing other continuous variables (eg, age, BMI SD score [SDS], cortisol excess measures) between those with and without remission. Categorical data (eg, sex, Tanner stage) were analyzed using the Fisher exact test. Comparisons of copeptin levels among the subgroups (DI, SIADH, neither) were carried out using mixed models and the Kruskal-Wallis test, as appropriate. Post hoc pairwise comparisons were adjusted for multiplicity using the Bonferroni correction, and as applicable, only corrected P values are reported. Mixed models for repeated measures also analyzed copeptin, serum sodium, and cortisol data for PODs 1 through 8. In addition, maximum likelihood estimation (GENMOD) procedures analyzed the effects of copeptin and serum sodium on the remission at follow-up. Correlation analyses were done with Spearman ρ. All analyses were tested for the potential confounding effects of age, sex, BMI SDS, and pubertal status, and were adjusted accordingly. For plasma copeptin reported as < 2.8 pmol/L, a value of 1.4 pmol/L (midpoint of 0 and 2.8 pmol/L) was used; sensitivity analyses repeated all relevant comparisons using the threshold limit of 2.8 pmol/L instead of 1.4 pmol/L. Odds ratios (OR) and 95% CIs, other magnitudes of the effect, data variability, and 2-sided P values provided the statistical evidence for the conclusions. Statistical analyses were performed in SAS version 9.4 software (SAS Institute, Inc, Cary, NC).
Results
Patient Characteristics
Forty-four adult and pediatric patients, aged 7 to 55 years (77.2% were < 18 years old), with CD were included in the study. The cohort included 28 female patients (64%), and the median BMI SDS was 2.2 (1.1-2.5). Thirty-four percent (15/44) had prior pituitary surgery (none within the prior 6 weeks). Seventy-five percent (33/44) had postoperative follow-up evaluations available, with median follow-up of 13.5 months (11.3-16.0). Of those 33 patients, 85% were determined to be in remission at follow-up. Comparing those in remission vs no remission, there was no difference in age, sex, BMI SDS, pubertal status (in pediatric ages only), preoperative measures of cortisol excess (UFC × ULN, PM serum cortisol, MN plasma ACTH, AM plasma ACTH), duration of follow-up, or development of DI or SIADH. There was a lower postoperative serum cortisol nadir in those in remission at follow-up compared with those not in remission at follow-up, as expected, because a postoperative serum cortisol < 5 μg/dL was included in defining remission status. Postoperatively, 8/44 (18%) developed DI, 13/44 (30%) developed SIADH, 4/44 (9%) developed both DI and SIADH, and 19/44 (43%) developed no water balance disorder (Table 1). There were no differences by remission status when assessing these subgroups (ie, DI, SIADH, and no water balance disorder) separately.
Table 1.Demographic and clinical characteristics of subjects
All subjects, n = 44 All subjects by remission status, n = 33 All subjects by remission status, excluding those with DI or SIADH, n = 13 Remission, n = 28 No remission, n = 5 P Remission,
n = 10No remission, n = 3 P Age, median (range), y 14.5 (7-55) 17.4 ± 10.7
14.5 (12.5-17.5)15.6 ± 13.2
11.0 (9.0-12.0)0.11 13.7 ± 3.1
14.0 (13.0-15.0)19.7 ± 16.8
11.0 (9.0-39.0)0.60a Sex
Female28 (64%) 22 (78.6%) 3 (60.0%) 0.57 9 (90.0%) 2 (66.7%) 0.42 BMI SDS 2.2 (1.1-2.5) 1.7 ± 1.0
2.0 (0.9-2.5)2.2 ± 0.4
2.2 (2.1-2.3)0.70 1.7 ± 1.1
2.0 (0.7-2.5)2.0 ± 0.4
2.1 (1.5-2.3)0.65a Pubertal status Female (n = 19) (n = 15) (n = 2) 0.51 (n = 8) (n = 1) 0.44 Tanner 1-2 6 4 (26.7%) 1 (50.0%) 3 (37.5%) 1 (25.0%) Tanner 3-5 13 11 (73.3%) 1 (50.0%) 5 (62.5%) 0 Male (n = 14) (n = 5) (n = 2) (n = 1) (n = 1) --- Testicular volume < 12, mL 10 4 (80.0%) 2 (10.00%) 1 (100.0%) 1 (100.0%) Testicular volume ≥ 12, mL 4 1 (20.0%) 0 1.0 0 0 Preoperative UFC ULN 3.3 (1.2-6.1) 4.9 ± 6.1
2.6 (1.0-7.6)3.2 ± 1.3
3.7 (3.0-3.9)0.70 7.2 ± 8.4
3.9 (1.8-9.1)3.8 ± 0.7
3.9 (3.0-4.4)0.93 Preoperative PM cortisol 11.9 (9.2-14.8) 13.3 ± 4.7
12.2 (9.2-16.8)10.8 ± 2.1
11.5 (9.0-11.6)0.30 13.3 ± 6.0
11.2 (8.4-16.5)11.1 ± 2.6
11.6 (8.3-13.6)0.57a Preoperative MN ACTH 43.4 (29.3-51.6) 44.2 ± 25.5
46.1 (27.6-50.5)40.9 ± 15.3
11.5 (9.0-11.6)0.74 36.6 ± 16.6
37.4 (29.1-48.8)34.0 ± 9.4
39.3 (23.1-39.5)0.67 Preoperative AM ACTH 44.6 (31.4-60.5) 46.9 ± 28.9
44.0 (29.8-56.2)48.6 ± 28.8
58.7 (21.7-60.5)0.84 35.2 ± 16.2
40.3 (28.0-44.0)45.4 ± 24.6
58.7 (17.0-60.5)0.41a Postoperative cortisol nadir 0.5 (0.5-0.5) 0.7 ± 0.7
0.5 (0.5-0.5)7.8 ± 6.6
5.2 (2.2-12.3)<0.001 0.6 ± 0.3
0.5 (0.5-0.5)8.1 ± 7.9
5.2 (2.1-17.0)0.003 Duration of follow-up 13.5 (11.3-16.0) 15.3 ± 7.9
14.0 (12.0-16.5)14.0 ± 13.0
11.0 (6.0-14.0)0.30 18.6 ± 11.2
15.5 (12.0-27.0)16.7 ± 17.2
11.0 (3.0-36.0)0.82a DI only 8 (18%) 7/8 (87.5%) 1/8 (12.5%) 0.91 --- --- --- SIADH only 13 (30%) 8/9 (88.9%) 1/9 (11.1%) Neither DI/SIADH 19 (43%) 10/13 (76.9%) 3/13 (23.1%) Both DI and SIADH 4 (9%) 3/3 (100%) 0/3 Demographic and clinical characteristics of all subjects (n = 44) with Cushing disease. Data are also presented by remission status for all subjects with postoperative follow-up (n = 33) and by remission status after excluding those who developed DI or SIADH postoperatively with postoperative follow-up (n = 13). Both median (IQR) and mean ± SD reported to allow for comparisons, with P value provided using appropriate testing depending on distribution of data sets. Data are mean ± SD, median (25th-75th IQR), or frequency (percentage) are reported, except for age, which is presented as median (range).
Abbreviations: AM, 7:30-8 PM; BMI, body mass index; DI, diabetes insipidus; IQR, interquartile range; MN, midnight; N/A, not applicable; SDS, SD score; SIADH, syndrome of inappropriate antidiuresis; UFC, urinary free cortisol; ULN, upper limit of normal. p-values below the threshold of 0.05 are in bold.
aP value indicates comparison using parametric testing, as appropriate for normally distributed data.
Preoperative copeptin levels were higher in males (7.0 pmol/L [5.1-9.6]) than in females (4.0 pmol/L [1.4-5.8], P = 0.004) (Fig. 1). Age was inversely correlated with preoperative copeptin (rs = -0.35, P = 0.030) and BMI SDS was positively correlated with preoperative copeptin (rs = 0.54, P < 0.001) (Fig. 2).
Figure 1.Preoperative plasma copeptin and sex. Preoperative plasma copeptin in all patients, comparing by sex. A higher preoperative plasma copeptin was found in males (7.0 pmol/L [5.1-9.6]) than in females (4.0 pmol/L [1.4-5.8], P = 0.004). Horizontal lines = median. Whiskers = 25th and 75th interquartile ranges.
![Preoperative plasma copeptin and sex. Preoperative plasma copeptin in all patients, comparing by sex. A higher preoperative plasma copeptin was found in males (7.0 pmol/L [5.1-9.6]) than in females (4.0 pmol/L [1.4-5.8], P = 0.004). Horizontal lines = median. Whiskers = 25th and 75th interquartile ranges.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jes/6/6/10.1210_jendso_bvac053/1/m_bvac053f0001.jpeg?Expires=1655299443&Signature=cGAP0Evman3rpz4yG5HWaDLcRC1jc1TJk93oOvoSu6VA1t1EWSDzSbl9EhdBf9JXp71MAF1oDWjp-6pLdnX31L06-LpLsjkSotr8DlUIvLNM9Z04l-gqSGf01AdXbLbpLDFsact6VqUeowhdAqNnh9zTcW0koOiQQHfFOhEUKieUoD1969Ae5bBh21~XnjFLQgGnckgxnVx3UCot7j~HS2iAi8ymdz0VDnNDzeooog4Youey2szlxX0O8LkEbCh0ZKP1evFH-tj6655-d9cYmGNUxBdO7SSIIcEnP9F29Lez~5JloMhqD~0RX2eL76VQ6iIWvd~wGdMmYQg~cIWFmw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA) Figure 2.
Figure 2.Preoperative plasma copeptin and BMI SDS. Association of preoperative plasma copeptin and BMI SDS in all patients. A BMI SDS was positively associated with a preoperative plasma copeptin (rs = 0.54, P < 0.001). Shaded area = 95% confidence interval.

Copeptin Before and After Transsphenoidal Surgery for CD
Among the 33 patients with postoperative follow-up, there was no difference in peak postoperative copeptin for patients in remission vs those not in remission (6.1 pmol/L [4.3-12.1] vs 7.3 pmol/L [5.4-8.4], P = 0.88). There was also no difference in the Δcopeptin for those in remission vs not in remission (2.3 pmol/L [-0.5 to 8.2] vs 0.1 pmol/L [-0.1 to 2.2], P = 0.46) (Fig. 3). Including all subjects, the mean preoperative copeptin was 5.6 pmol/L (±3.4). For patients with follow-up, there was no difference in preoperative copeptin for those in remission (4.8 pmol/L [±2.9]) vs no remission (6.0 pmol/L [±2.0], P = 0.47). POD 1 plasma copeptin ranged from < 2.8 to 11.3 pmol/L.
Figure 3.(A) Peak postoperative plasma copeptin in all patients, comparing those in remission with no remission (6.1 pmol/L [4.3-12.1] vs 7.3 pmol/L [5.4-8.4], P = 0.88). (B) ΔCopeptin (preoperative plasma copeptin subtracted from postoperative peak plasma copeptin) in all patients, comparing those in remission with no remission (2.3 pmol/L [-0.5 to 8.2] vs 0.1 pmol/L [-0.1 to 2.2], P = 0.46). Horizontal lines = median. Whiskers = 25th and 75th interquartile ranges.
![(A) Peak postoperative plasma copeptin in all patients, comparing those in remission with no remission (6.1 pmol/L [4.3-12.1] vs 7.3 pmol/L [5.4-8.4], P = 0.88). (B) ΔCopeptin (preoperative plasma copeptin subtracted from postoperative peak plasma copeptin) in all patients, comparing those in remission with no remission (2.3 pmol/L [-0.5 to 8.2] vs 0.1 pmol/L [-0.1 to 2.2], P = 0.46). Horizontal lines = median. Whiskers = 25th and 75th interquartile ranges.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jes/6/6/10.1210_jendso_bvac053/1/m_bvac053f0003.jpeg?Expires=1655299443&Signature=yW5PElkusuxtGjvn1AjQypt5XCd0KjXFXHAMhFsaWHhKnzKTixeryNbeCy109hsnlgnxVujb2SpQX04MH6oHBT2O6xo50vxQEcQgcC~ioyFJtMyk5-F24MkV0Szwr6~XIVOqNbs5adU91Y6AloIq2sUwxDPPrO8HHFwazLMXeZp8716bss98UtRM-SnpRYk~JoL4mfSg1jtwghv-1Jim5a3BoZEd6z7PJPx-RNz5jAsUeLvpwOW~4ylC6vEKs~3Wj4bc6VDPLA56TmowuV6I5NDgTiIErbtwKfRVn82d2216XXf72uaQc8rjp8pVNvLc6xrAneMt7C-sVuhxAPMEuQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
When those who developed DI or SIADH were excluded, there was no difference in peak postoperative copeptin in those in remission vs no remission (10.2 pmol/L [6.9-21.0] vs 5.4 pmol/L [4.6-7.3], P = 0.20). However, because the distribution of the peak postoperative copeptins was borderline normally distributed, parametric testing was also completed for this analysis, which showed a higher peak postoperative copeptin in remission (14.6 pmol/L [±10.9]) vs no remission (5.8 [±1.4], P = 0.03). There was no difference in the Δcopeptin for those in remission vs not in remission (5.1 pmol/L [0.3-19.5] vs 1.1 pmol/L [-0.1 to 2.2], P = 0.39) (Fig. 4). Preoperative copeptin was not different for those in remission (4.7 pmol/L [±2.4]) vs no remission (4.9 pmol/L [±20.3], P = 0.91). There was no association between serum cortisol and plasma copeptin over time postoperatively (Fig. 5).
Figure 4.(A) Peak postoperative plasma copeptin excluding those who developed DI or SIADH, comparing those in remission with no remission (10.2 pmol/L [6.9-21.0] vs 5.4 pmol/L [4.6-7.3], P = 0.20). (B) ΔCopeptin (preoperative plasma copeptin subtracted from postoperative peak plasma copeptin) excluding those who developed DI or SIADH, comparing those in remission with no remission (5.1 pmol/L [0.3-19.5] vs 1.1 pmol/L [-0.1 to 2.2], P = 0.39). Horizontal lines = median. Whiskers = 25th and 75th interquartile ranges.
![(A) Peak postoperative plasma copeptin excluding those who developed DI or SIADH, comparing those in remission with no remission (10.2 pmol/L [6.9-21.0] vs 5.4 pmol/L [4.6-7.3], P = 0.20). (B) ΔCopeptin (preoperative plasma copeptin subtracted from postoperative peak plasma copeptin) excluding those who developed DI or SIADH, comparing those in remission with no remission (5.1 pmol/L [0.3-19.5] vs 1.1 pmol/L [-0.1 to 2.2], P = 0.39). Horizontal lines = median. Whiskers = 25th and 75th interquartile ranges.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jes/6/6/10.1210_jendso_bvac053/1/m_bvac053f0004.jpeg?Expires=1655299443&Signature=IQ4ivUkf6XLkyDLGryzJjbHj9DhLWOhbnOr5ofRMRJY-NQbXQfMfEkvlD0q-zYHnLQMu~mnVWBdDsOxYYc-QiLAwqGX7qwUiOgqYy1r1U7XwmOHMASKP6EhdEpkdL7cxyof~1nGKjnCvvQMoP-r81P~Ce1N0Behb2CR8LrCfPYayWxRY0B5nPis3twtddA2vZxdH4iZpVUkqB~CA5BGsMynR62A~~XsFIQfB8kbnEPipNOWvd1A0-GzS-O520eo6TrPfLV3iqJ3wCo7oJ4Z42ziIcgxplWOX5EuaMU59tRTA6lgmT8GKFTg8yhpLgC-wevkk2LCSTWSk9Y3GMrKptQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA) Figure 5.
Figure 5.Plasma copeptin and serum cortisol vs postoperative day for patients who did not develop DI or SIADH. Plasma copeptin (indicated by closed circle) and serum cortisol (indicated by “x”). Results shown as (median, 95% CI).

All analyses here were repeated adjusting for serum sodium, and there were no differences by remission status for preoperative, peak postoperative, or Δcopeptin for all subjects or after excluding those who developed a water balance disorder (data not shown).
Copeptin and Water Balance Disorders
As expected, peak postoperative copeptin appeared to be different among patients who developed DI, SIADH, and those without any fluid balance disorder (P = 0.029), whereas patients with DI had lower median peak postoperative copeptin (4.4 pmol/L [2.4-6.9]) than those who developed no fluid abnormality (10.0 pmol/L [5.4-16.5], P = 0.04), the statistical difference was not present after correction for multiple comparisons (P = 0.13). Peak postoperative copeptin of patients with SIADH was 9.4 pmol/L (6.5-10.4) and did not differ from patients with DI (P = 0.32) or those with no fluid abnormality (P = 1.0). There was a difference in Δcopeptin levels among these subgroups (overall P = 0.043), which appeared to be driven by the lower Δcopeptin in those who developed DI (-1.2 pmol/L [-2.6 to 0.1]) vs in those with neither DI or SIADH (3.1 pmol/L [0-9.6], P = 0.05). However, this pairwise comparison did not reach statistical significance, even before correction for multiple comparisons (P = 0.16) (Fig. 6). Preoperative copeptin levels were also not different among the subgroups (P = 0.54).
Figure 6.(A) Peak postoperative plasma copeptin, comparing those who developed DI, SIADH, or neither (P = 0.029 for comparison of all 3 groups). (B) ∆ Copeptin (preoperative plasma copeptin subtracted from postoperative peak plasma copeptin), comparing those who developed DI, SIADH, or neither (P = 0.043 for comparison of all 3 groups). Horizontal lines = median. Whiskers = 25th and 75th interquartile ranges. Top brackets = pairwise comparisons. P values presented are after Bonferroni correction for multiple comparisons.

Association of Sodium and Copeptin
Longitudinal data, adjusting for subgroups (ie, DI, SIADH, neither), were analyzed. As expected, there was a group difference (P = 0.003) in serum sodium over time (all DI was missing preoperative serum sodium), with the difference being driven by DI vs SIADH (P = 0.007), and SIADH vs neither (P = 0.012). There was no group difference in plasma copeptin over POD by water balance status (P = 0.16) over time (Fig. 7). There was also no effect by remission status at 3 to 6 months for either serum sodium or plasma copeptin.
Figure 7.(A) Serum sodium and (B) plasma copeptin by POD and water balance status longitudinal data, adjusting for subgroups (ie, DI, SIADH, neither). Data points at point 0 on the x-axis indicate preoperative values. As expected, there was a group difference (P = 0.003) in serum sodium over time (all with DI were missing preoperative serum sodium), with the difference being driven by DI vs SIADH (P = 0.007), and SIADH vs neither (P = 0.012). There was no group difference in plasma copeptin over POD by water balance status (P = 0.16) over time.

Higher serum sodium levels from PODs 1 through 8 itself decreased the odds of remission (OR, 0.56; 95% CI, 0.42-0.73; P < 0.001) in all CD patients. Copeptin levels from these repeated measures adjusting for serum sodium did not correlate with remission status at 3 to 6 months’ follow-up (P = 0.38). There were no differences in preoperative, peak postoperative, or delta sodium levels by remission vs no remission in all patients and in those with no water balance disorders.
Discussion
AVP and CRH act synergistically to stimulate the secretion of ACTH and ultimately cortisol [1, 2], and there is evidence that glucocorticoids act by way of negative feedback to suppress AVP secretion [10, 11-20]. Therefore, we hypothesized that a greater postoperative increase in plasma copeptin in those with CD in remission after TSS because of resolution of hypercortisolemia and resultant hypocortisolemia, compared with those not in remission with persistent hypercortisolemia and continued negative feedback, would be observed. Although a clear difference in peak postoperative and Δcopeptin was not observed in this study, a higher peak postoperative copeptin was found in those in remission after excluding those who developed DI/SIADH when analyzing this comparison with parametric testing, and it is possible that we did not have the power to detect a difference by nonparametric testing, given our small sample size. Therefore, postoperative plasma copeptin may be a useful early marker to predict remission of CD after TSS. The utility of this test may be limited to those who do not develop water balance disorders postoperatively. If a true increase in copeptin occurs for those in remission after treatment of CD, it is possible that this could be due to the removal of negative feedback from cortisol excess on pre-pro-AVP secretion, as hypothesized in this study. However, it is also possible that other factors may contribute to an increase in copeptin postoperatively, including from the stress response of surgery and postoperative hypocortisolism and resultant stimulation of pre-pro-AVP secretion from these physical stressors and/or from unrecognized SIADH.
It was anticipated that more severe hypercortisolism to be negatively correlated with preoperative plasma copeptin because of greater negative feedback on AVP. However, no association was found between preoperative plasma copeptin and markers of severity of hypercortisolism (MN cortisol, AM ACTH, UFC × ULN) in this study. Similarly, we would expect that the preoperative plasma copeptin would be lower compared with healthy individuals. However, comparisons of healthy individuals may be difficult because the fluid and osmolality status at the time of the sample could influence the plasma copeptin, and depending on those factors, copeptin could be appropriately low. A healthy control group with whom to compare the preoperative values was not available for this study, and the thirsted state was not standardized for the preoperative copeptin measurements. Future studies could be considered to determine if preoperative plasma copeptin is lower in patients with CD, or other forms of CS, compared with healthy subjects, with all subjects thirsted for an equivalent period. Further, if preoperative plasma copeptin is found to be lower in thirsted subjects with CS than a thirsted healthy control group, the plasma copeptin could potentially be a diagnostic test to lend support for or against the diagnosis of endogenous CS.
In the comparisons of those who developed DI, SIADH, or neither, no difference was found in the Δcopeptin. Peak copeptin was lower in DI compared with those without DI or SIADH (but not different from SIADH). Again, it is possible that there is a lower peak postoperative copeptin and change in copeptin in those with DI, but we may not have had the power to detect this in all of our analyses. These comparisons of copeptin among those with or without water balance disorders postoperatively are somewhat consistent with a prior study showing postoperative copeptin as a good predictor of development of DI, in which a plasma copeptin < 2.5 pmol/L measured on POD 0 accurately identified those who developed DI, and plasma copeptin > 30 pmol/L ruled out the development of DI postoperatively [29]. In the current study, 3 of 6 subjects with DI had a POD 1 plasma copeptin < 2.5 pmol/L, and none had a POD 1 plasma copeptin > 30 pmol/L. However, the study by Winzeler et al found that copeptin measured on POD 0 (within 12 hours after surgery) had the greatest predictive value, and POD 0 plasma copeptin was not available in our study. Further, we used the preoperative, peak, and delta plasma copeptin for analyses, so the early low copeptin levels may not have been captured in our data and analyses.
Additionally, this study revealed that increasing levels of serum sodium have lower odds of remission. Those who have an ACTH-producing adenoma that is not identified by magnetic resonance imaging and visual inspection intraoperatively have lower rates of remission and are more likely to have greater manipulation of the pituitary gland intraoperatively [32-36], and the latter may result in greater damage to the pituitary stalk or posterior pituitary, increasing the risk for development of DI and resultant hypernatremia.
A higher preoperative copeptin was associated with male sex and increasing BMI SDS. Increasing preoperative copeptin was also found in pubertal boys compared with pubertal girls, with no difference in copeptin between prepubertal boys and girls. It is particularly interesting to note that these associations were only in the preoperative plasma copeptin levels, but not the postoperative peak copeptin or Δcopeptin. Because the association of higher plasma in adult males and pubertal males in comparison to adult females and pubertal females, respectively, have been reported by others [26, 37-40], it raises the question of a change in the association of sex and BMI with plasma copeptin in the postoperative state. An effect of BMI or sex was not found by remission status, so it does not seem that the postoperative hypocortisolemic state for those in remission could explain this loss of association. However, this study may not have been powered to detect this.
Strengths of this study include the prospective nature of the study. Further, this is the first study assessing the utility of copeptin to predict remission after treatment of CD. Limitations of this study include the small sample size because of the rarity of the condition, difficulty in clinically diagnosing DI and SIADH, potential effect of post-TSS fluid balance disorders (particularly for those who may have developed transient partial DI or transient SIADH), lack of long-term follow-up, lack of any postoperative follow-up in 11 of the 44 total subjects, as well the observational nature of the study. Further, it is possible that pubertal status, sex, and BMI may have affected copeptin levels, which may have not been consistently detected because of lack of power. Lack of data on the timing of hydrocortisone replacement is an additional limitation of this study because postoperative glucocorticoid replacement could affect AVP secretion via negative feedback. Additional studies are needed to assess to further assess the role of vasopressin and measurement of copeptin in patients before and after treatment of CD.
A clear difference in peak postoperative plasma copeptin as an early marker to predict remission of CD after TSS was not found. Further studies with larger sample sizes are needed to further evaluate postoperative plasma copeptin as an early marker to predict remission of CD, though the utility of this test may be limited to those who do not develop water balance disorders postoperatively. Future studies comparing copeptin levels before and after treatment of adrenal CS would be of particular interest because this would minimize the risk of postoperative DI or SIADH which also influence copeptin levels. Additionally, comparison of thirsted preoperative plasma copeptin in those with endogenous CS and thirsted plasma copeptin in healthy controls could potentially provide evidence of whether or not preoperative plasma copeptin is lower in patients with CD, or other forms of CS, compared with healthy subjects. Further, if this is found to be true, it could potentially be a diagnostic test to lend support for or against endogenous CS.
Abbreviations
-
AVP
arginine vasopressin
-
BMI
body mass index
-
CD
Cushing disease
-
CS
Cushing syndrome
-
DI
diabetes insipidus
-
HPA
hypothalamic-pituitary-adrenal
-
IQR
interquartile range
-
MN
midnight
-
OR
odds ratio
-
POD
postoperative day
-
SDS
SD score
-
SIADH
syndrome of inappropriate antidiuresis
-
TSS
transsphenoidal surgery
-
UFC
urinary free cortisol
-
ULN
upper limit of normal
Acknowledgments
The authors thank the patients and their families for participating in this study.
Funding
This work was supported by the Intramural Research Program, Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD), National Institutes of Health.
Disclosures
C.A.S. holds patents on technologies involving PRKAR1A, PDE11A, GPR101, and related genes, and his laboratory has received research funding support by Pfizer Inc. for investigations unrelated to this project. C.A.S. is associated with the following pharmaceutical companies: ELPEN, Inc., H. Lunbeck A/S, and Sync. Inc.
Clinical Trial Information
ClinicalTrials.gov registration no. NCT00001595 (registered November 4, 1999).
Data Availability
Some or all datasets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.
Published by Oxford University Press on behalf of the Endocrine Society 2022.This work is written by (a) US Government employee(s) and is in the public domain in the US.-
1
-
-
Research opportunity for Human Growth Hormone Deficiency caregivers of adolescent patients.
This is a 75 min web-assisted phone interview, and the compensation is $125.
Please sign up at the link below to receive an email invite to the survey.
-
1
-
-
Abstract
ObjectiveWe aimed to perform a systematic review and meta-analysis of all-cause and cause-specific mortality of patients with benign endogenous Cushing's syndrome (CS).
MethodsThe protocol was registered in PROSPERO (CRD42017067530). PubMed, EMBASE, CINHAL, Web of Science and Cochrane Central searches were undertaken from inception to January 2021. Outcomes were the standardized mortality ratio (SMR), proportion and cause of deaths. The I 2 test, subgroup analysis and meta-regression were used to assess heterogeneity across studies.
ResultsSMR was reported in 14 articles including 3,691 patients (13 Cushing's disease (CD) and 7 adrenal CS (ACS) cohorts). Overall SMR was 3.0 (95%CI 2.3-3.9; I 2=80.5%) for all CS, 2.8 (95%CI 2.1-3.7 I 2=81.2%) for CD and 3.3 (95%CI 0.5-6.6; I 2=77.9%) for ACS. Proportion of deaths, reported in 87 articles including 19,181 CS patients (53 CD, 24 ACS, and 20 combined CS cohorts) was 0.05 (95%CI 0.03, 0.06) for all CS subtypes with meta-regression analysis revealing no differences between CS subtypes (P=0.052). The proportion of deaths was 0.1 (10%) in articles published before 2000 and 0.03 (3%) in 2000 until the last search for CS (P<0.001), CD (p<0.001), and ACS (P=0.01). The causes of death were atherosclerotic diseases and thromboembolism (43.4%), infection (12.7%), malignancy (10.6%), active disease (3.5%), adrenal insufficiency (3.0%), and suicide (2.2%).
Despite improved outcomes in recent years, increased mortality from CS persists. The causes of death highlight the need to prevent and manage co-morbidities in addition to treating hypercortisolism.
 Accepted manuscripts
Accepted manuscripts are PDF versions of the author’s final manuscript, as accepted for publication by the journal but prior to copyediting or typesetting. They can be cited using the author(s), article title, journal title, year of online publication, and DOI. They will be replaced by the final typeset articles, which may therefore contain changes. The DOI will remain the same throughout.This content is only available as a PDF.© The Author(s) 2022. Published by Oxford University Press on behalf of the Endocrine Society.This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse, distribution, and reproduction in any medium, provided the original work is properly cited.
Accepted manuscripts
Accepted manuscripts are PDF versions of the author’s final manuscript, as accepted for publication by the journal but prior to copyediting or typesetting. They can be cited using the author(s), article title, journal title, year of online publication, and DOI. They will be replaced by the final typeset articles, which may therefore contain changes. The DOI will remain the same throughout.This content is only available as a PDF.© The Author(s) 2022. Published by Oxford University Press on behalf of the Endocrine Society.This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse, distribution, and reproduction in any medium, provided the original work is properly cited.-
1
-
-
Osilodrostat is associated with rapid normalization of mean urinary free cortisol (mUFC) excretion in patients with Cushing disease and has a favorable safety profile, according to the results of a study published in the Journal of Clinical Endocrinology & Metabolism.
The phase 3 LINC-4 study (ClinicalTrials.gov Identifier: NCT02697734) evaluated the safety and efficacy of osilodrostat, a potent, orally available 11β-hydroxylase inhibitor, compared with placebo in patients with Cushing disease.
The trial, which was conducted at 40 centers in 14 countries, included a 12-week, randomized, double-blind, placebo-controlled period that was followed by a 36-week, open-label osilodrostat treatment period with an optional extension.
Eligible patients were aged 18 to 75 years with a confirmed diagnosis of persistent or recurrent Cushing disease after pituitary surgery and/or irradiation or de novo disease, as well as an mUFC level greater than 1.3 times the upper limit of normal (ULN). The patients were randomly assigned 2:1 to osilodrostat 2 mg twice daily or matching placebo, stratified by prior pituitary irradiation.
The primary endpoint was the proportion of patients who achieved mUFC ≤ULN at week 12. The key secondary endpoint was the proportion of patients who achieved mUFC ≤ULN at week 36.
A total of 73 patients (median age, 39.0 years; 83.6% women) were randomly assigned to either osilodrostat (n=48) or placebo (n=25) and received at least 1 study drug dose from November 2016 to March 2019.
The participants had a median (interquartile range [IQR]) time since diagnosis of Cushing disease of 67.4 (26.4-93.8) months. The median treatment duration in the randomized, placebo-controlled period was 12.0 weeks in both the osilodrostat group (IQR, 2.0-13.0 weeks) and the placebo group (IQR, 11.7-13.7 weeks).
The proportion of patients who achieved mUFC ≤ULN (≤138 nmol/24 h) at week 12 was significantly increased in those who received osilodrostat (n=37, 77.1%) vs those who received placebo (n=2, 8.0%), with an estimated odds ratio of 43.4 (95% CI, 7.1-343.2) in favor of osilodrostat (P <.0001).
A total of 59 patients (80.8%; 95% CI, 69.9-89.1) also achieved the key secondary endpoint of mUFC ≤ULN at week 36, after 24 weeks of open-label osilodrostat.
The most frequently occurring adverse events in the placebo-controlled period in the osilodrostat and placebo groups, respectively, were decreased appetite (37.5% vs 16.0%), arthralgia (35.4% vs 8.0%), nausea (31.3% vs 12.0%), and fatigue (25.0% vs 16.0%).
A potential study limitation is that although osilodrostat exposure was greater than 1 year among the participants, some adverse effects may take longer to be observed.
“This randomized, placebo-controlled trial demonstrates that osilodrostat is a highly effective treatment for Cushing disease, normalizing UFC excretion in 77% of patients after 12 weeks’ treatment,” stated the investigators. “Cortisol reductions were maintained throughout 48 weeks of treatment and were accompanied by improvements in clinical signs of hypercortisolism and quality of life. The safety profile was favorable.”
Disclosure: This study was funded by Novartis Pharma AG. Some of the study authors declared affiliations with biotech, pharmaceutical, and/or device companies. Please see the original reference for a full list of disclosures.
Reference
Gadelha M, Bex M, Feelders RA, et al. Randomized trial of osilodrostat for the treatment of Cushing’s disease. J Clin Endocrinol Metab. Published online March 23, 2022. doi:10.1210/clinem/dgac178
-
1
-
-
The popular website "How Stuff Work"s is doing a survey of all kinds of diseases and Cushing's is one of them!
Share your information and help get the word out to the world in general.
(I'm MaryO there, too and I shared about my pituitary surgery and its aftermath. I hope this info helps someone else like these boards and related websites have)
The questionnaire is here: https://stuff.health/s/
u0A9djA5
Together, we’ll figure out which treatments work best for Cushing's syndrome.
-
1
-
-
Abstract
Background
Neuroendocrine tumors can cause ectopic Cushing syndrome, and most patients have metastatic disease at diagnosis. We identified risk factors for outcome, evaluated ectopic Cushing syndrome management, and explored the role of bilateral adrenalectomy in this population.Methods
This was a retrospective study including patients with diagnosis of ectopic Cushing Syndrome secondary to neuroendocrine tumors with adrenocorticotropic hormone secretion treated at our quaternary referral center over a 40-year period (1980–2020).Results
Seventy-six patients were included. Mean age at diagnosis was 46.3 ± 15.8 years. Most patients (N = 61, 80%) had metastases at ectopic Cushing syndrome diagnosis. Average follow-up was 2.9 ± 3.7 years (range, 4 months–17.2 years). Patients with neuroendocrine tumors before ectopic Cushing syndrome had more frequent metastatic disease and resistant ectopic Cushing syndrome. Patients with de novo hyperglycemia, poor neuroendocrine tumor differentiation, and metastatic disease had worse survival. Of those with nonmetastatic disease, 8 (53%) had ectopic Cushing syndrome resolution after neuroendocrine tumor resection, 3 (20%) were medically controlled, and 4 (27%) underwent bilateral adrenalectomy. In patients with metastatic neuroendocrine tumors, hypercortisolism was initially medically managed in 92%, 3% underwent immediate bilateral adrenalectomy, 2% had control after primary neuroendocrine tumor debulking, and 2% were lost to follow-up. Medical treatment resulted in hormonal control in 7 (13%) patients. Of the 49 patients with metastatic disease and medically resistant ectopic Cushing syndrome, 23 ultimately had bilateral adrenalectomy with ectopic Cushing syndrome cure in all.Conclusion
Patients with neuroendocrine tumors before ectopic Cushing syndrome development were more likely metastatic and had worse survival. De novo hyperglycemia and poor neuroendocrine tumor differentiation were predictive of worse prognosis. Medical control of hypercortisolism is difficult to achieve in patients with neuroendocrine tumors–ectopic Cushing syndrome. Well-selected patients may benefit from bilateral adrenalectomy early in the treatment algorithm, and multidisciplinary management is essential in this complex disease.Graphical abstract

More information at https://www.surgjournal.com/article/S0039-6060(22)00158-1/fulltext
-
1
-
-
Today is the final day of the Cushing’s Awareness Challenge and I wanted to leave you with this word of advice…

To that end, I’m saving some of what I know for future blog posts, maybe even another Cushing’s Awareness Challenge next year. Possibly this has become a tradition.
I am amazed at how well this Challenge went this year, giving that we’re all Cushies who are dealing with so much. I hope that some folks outside the Cushing’s community read these posts and learned a little more about us and what we go through.
So, tomorrow, I’ll go back to posting the regular Cushing’s stuff on this blog – after all, it does have Cushing’s in its name!
I am trying to get away from always reading, writing, breathing Cushing’s, and trying to celebrate the good things in my life, not just the testing, the surgery, the endless doctors.
If you’re interested, I have other blogs about traveling, friends, fun stuff and trying to live a good life, finally. Those are listed in the right sidebar of this blog, past the Categories and before the Tags.
Meanwhile...
http://maryoblog.files.wordpress.com/2011/07/time-for-me-scaled500.jpg?w=314&h=283&h=283Choose wisely...
http://cushieblog.files.wordpress.com/2012/04/maryo-colorful-zebra1.gif
-
1
-
-
Abstract
Cushing’s syndrome (CS) secondary to ectopic adrenocorticotrophic hormone (ACTH)-producing prostate cancer is rare with less than 50 cases reported. The diagnosis can be challenging due to atypical and variable clinical presentations of this uncommon source of ectopic ACTH secretion. We report a case of Cushing’s syndrome secondary to prostate adenocarcinoma who presented with symptoms of severe hypercortisolism with recurrent hypokalaemia, limb oedema, limb weakness, and sepsis. He presented with severe hypokalaemia and metabolic alkalosis (potassium 2.5 mmol/L and bicarbonate 36 mmol/L), with elevated 8 am cortisol 1229 nmol/L. ACTH-dependent Cushing’s syndrome was diagnosed with inappropriately normal ACTH 57.4 ng/L, significantly elevated 24-hour urine free cortisol and unsuppressed cortisol after 1 mg low-dose, 2-day low-dose, and 8 mg high-dose dexamethasone suppression tests. 68Ga-DOTANOC PET/CT showed an increase in DOTANOC avidity in the prostate gland, and his prostate biopsy specimen was stained positive for ACTH and markers for neuroendocrine differentiation. He was started on ketoconazole, which was switched to IV octreotide in view of liver dysfunction from hepatic metastases. He eventually succumbed to the disease after 3 months of his diagnosis. It is imperative to recognize prostate carcinoma as a source of ectopic ACTH secretion as it is associated with poor clinical outcomes, and the diagnosis can be missed due to atypical clinical presentations.
1. Introduction
Ectopic secretion of adrenocorticotropic hormone (ACTH) is responsible for approximately 10–20% of all causes of Cushing syndrome [1]. The classic sources of ectopic ACTH secretion include bronchial carcinoid tumours, small cell lung carcinoma, thymoma, medullary thyroid carcinoma (MTC), gastroenteropancreatic neuroendocrine tumours (NET), and phaeochromocytomas [2]. Ectopic adrenocorticotropic syndrome (EAS) is diagnostically challenging due to its variable clinical manifestations; however, prompt recognition and treatment is critical. Ectopic ACTH production from prostate carcinoma is rare, and there are less than 50 cases published to date. Here, we report a case of ectopic Cushing’s syndrome secondary to prostate adenocarcinoma who did not present with the typical physical features of Cushing’s syndrome, but instead with features of severe hypercortisolism such as hypokalaemia, oedema, and sepsis.
2. Case Presentation
A 61-year-old male presented to our institution with recurrent hypokalaemia, lower limb weakness, and oedema. He had a history of recently diagnosed metastatic prostate adenocarcinoma, for which he was started on leuprolide and finasteride. Other medical history includes poorly controlled diabetes mellitus and hypertension of 1-year duration. He presented with hypokalaemia of 2.7 mmol/L associated with bilateral lower limb oedema and weakness, initially attributed to the intake of complementary medicine, which resolved with potassium supplementation and cessation of the complementary medicine. One month later, he was readmitted for refractory hypokalaemia of 2.5 mmol/L and progression of the lower limb weakness and oedema. On examination, his blood pressure (BP) was 121/78 mmHg, and body mass index (BMI) was 24 kg/m2. He had no Cushingoid features of rounded and plethoric facies, supraclavicular or dorsocervical fat pad, ecchymoses, and no purple striae on the abdominal examination. He had mild bilateral lower limb proximal weakness and oedema.
His initial laboratory findings of severe hypokalaemia with metabolic alkalosis (potassium 2.5 mmol/L and bicarbonate 36 mmol/L), raised 24-hour urine potassium (86 mmol/L), suppressed plasma renin activity and aldosterone, central hypothyroidism, and elevated morning serum cortisol (1229 nmol/L) (Table 1) raised the suspicion for endogenous hypercortisolism. Furthermore, hormonal evaluations confirmed ACTH-dependent Cushing’s syndrome with inappropriately normal ACTH (56 ng/L) and failure of cortisol suppression after 1 mg low-dose, 2-day low-dose, and 8 mg high-dose dexamethasone suppression tests (Table 2). His 24-hour urine free cortisol (UFC) was significantly elevated at 20475 (59–413) nmol/day.
To identify the source of excessive cortisol secretion, magnetic resonance imaging (MRI) of the pituitary fossa and computed tomography (CT) of the thorax, abdomen, and pelvis were performed. Pituitary MRI was unremarkable, and CT scan showed the known prostate lesion with extensive liver, lymph nodes, and bone metastases (Figure 1). To confirm that the prostate cancer was the source of ectopic ACTH production, gallium-68 labelled somatostatin receptor positron emission tomography (PET)/CT (68Ga-DOTANOC) was done, which showed an increased DOTANOC avidity in the inferior aspect of the prostate gland (Figure 2). Immunohistochemical staining of his prostate biopsy specimen was requested, and it stained positive for ACTH and markers of neuroendocrine differentiation (synaptophysin and CD 56) (Figures 3 and 4), establishing the diagnosis of EAS secondary to prostate cancer.
The patient was started on potassium chloride 3.6 g 3 times daily and spironolactone 25 mg once daily with normalisation of serum potassium. His BP was controlled with the addition of lisinopril and terazosin to spironolactone and ketoconazole, and his blood glucose was well controlled with metformin and sitagliptin. To manage the hypercortisolism, he was treated with ketoconazole 400 mg twice daily with an initial improvement of serum cortisol from 2048 nmol/L to 849 nmol/L (Figure 5). Systemic platinum and etoposide-based chemotherapy was recommended for the treatment of his prostate cancer after a multidisciplinary discussion, but it was delayed due to severe bacterial and viral infection. With the development of liver dysfunction, ketoconazole was switched to intravenous octreotide 100 mcg three times daily as metyrapone was not readily available in our country. However, the efficacy was suboptimal with marginal reduction of serum cortisol from 3580 nmol/L to 3329 nmol/L (Figure 5). The patient continued to deteriorate and was deemed to be medically unfit for chemotherapy or bilateral adrenalectomy. He was referred to palliative care services, and he eventually demised due to cancer progression within 3 months of his diagnosis.
3. Discussion
Ectopic ACTH secretion is an uncommon cause of Cushing’s syndrome accounting for approximately 9–18% of the patients with Cushing’s syndrome [3]. Clinical presentation is highly variable depending on the aggressiveness of the underlying malignancy, but patients typically present with symptoms of severe hypercortisolism such as hypokalaemiaa, oedema, and proximal weakness which were the presenting complaints of our patient [4]. The classical symptoms of Cushing’s syndrome are frequently absent due to the rapid clinic onset resulting in diagnostic delay [5].
Prompt diagnosis and localisation of the source of ectopic ACTH secretion are crucial due to the urgent need for treatment initiation. The usual sources include small cell lung carcinoma, bronchial carcinoid, medullary thyroid carcinoma, thymic carcinoid, and pheochromocytoma. CT of the thorax, abdomen, and pelvis should be the first-line imaging modality, and its sensitivity varies with the type of tumour ranging from 77% to 85% [6]. Functional imaging such as 18-fluorodeoxyglucose-PET and gallium-68 labelled somatostatin receptor PET/CT can be useful in localising the source of occult EAS, determining the neuroendocrine nature of the tumour or staging the underlying malignancy [3, 6]. As prostate cancer is an unusual cause of EAS, we proceeded with 68Ga-DOTANOC PET/CT in our patient to localise the source of ectopic ACTH production.
The goals of management in EAS include treating the hormonal excess and the underlying neoplasm as well as managing the complications secondary to hypercortisolism [3]. Prompt management of the cortisol excess is paramount as complications such as hyperglycaemia, hypertension, hypokalaemia, pulmonary embolism, sepsis, and psychosis can develop especially when UFC is more than 5 times the upper limit of normal [3]. Ideally, surgical resection is the first-line management, but this may not be feasible in metastatic, advanced, or occult diseases.
Pharmacological agents are frequently required with steroidogenesis inhibitors such as ketoconazole and metyrapone, which reduce cortisol production effectively and rapidly [3, 6], the main drawback of ketoconazole being its hepatic toxicity. The efficacy of ketoconazole is reported to be 44%, metyrapone 50–75%, and ketoconazole-metyrapone combination therapy 73% [3, 7]. Mitotane, typically used in adrenocortical carcinoma, is effective in controlling cortisol excess but has a slow onset of action [3, 8]. Etomidate infusion can be used for short-term rapid control of severe symptomatic hypercortisolism and can serve as a bridge to definitive therapy [9]. Mifepristone, a glucocorticoid receptor antagonist, is indicated mainly in difficult to control hyperglycaemia secondary to hypercortisolism [8]. Somatostatin analogue has been proposed as a possible pharmacological therapy due to the expression of somatostatin receptors by ACTH secreting tumours [8, 10]. Bilateral adrenalectomy should be considered in patients with severe symptomatic hypercortisolism and life-threatening complications who cannot be optimally managed with medical therapies, especially in patients with occult EAS or metastatic disease [3, 8]. Bilateral adrenalectomy results in immediate improvement in cortisol levels and symptoms secondary to hypercortisolism [11]. However, surgical complications, morbidity, and mortality are high in patients with uncontrolled hypercortisolism [8], and our patient was deemed by his oncologist and surgeon to have too high a risk for bilateral adrenalectomy. For the treatment of prostate carcinoma, platinum and etoposide-based chemotherapies have been used, but their efficacy is limited with a median survival of 7.5 months [4, 12]. The side effects of chemotherapy can be severe with an enhanced risk of infection due to both cortisol and chemotherapy-mediated immunosuppression. Prompt control of hypercortisolism prior to chemotherapy and surgical procedure is strongly suggested to attenuate life-threatening complications such as infection, thrombosis, and bleeding with chemotherapy or surgery as well as to improve prognosis [3, 13].
There are rare reports of ectopic ACTH secretion from prostate carcinoma. These tumours were predominantly of small cell or mixed cell type, and pure adenocarcinoma with neuroendocrine differentiation are less common [4, 5]. There is a strong correlation between the prognosis and the types of malignancy in patients with EAS, and patients with prostate carcinoma have a poor prognosis [4]. These patients had metastatic disease at presentation, and the median survival was weeks to months despite medical treatment, chemotherapy, and even bilateral adrenalectomy [4], as seen with our patient who passed away within 3 months of his diagnosis.
In conclusion, adenocarcinoma of the prostate is a rare cause of EAS. The diagnosis and management are complex and challenging requiring specialised expertise with multidisciplinary involvement. The presentation can be atypical, and it is imperative to suspect and recognise prostate carcinoma as a source of ectopic ACTH secretion. Prompt initiation of treatment is important, as it is a rapidly progressive and aggressive disease associated with intense hypercortisolism resulting in high rates of mortality and morbidity.
Data Availability
The data used to support the findings of this study are included within the article.
Conflicts of Interest
The authors declare that there are no conflicts of interest.
Acknowledgments
The authors would like to thank the Pathology Department of Changi General Hospital for their contribution to this case.
References
- I. Ilias, D. J. Torpy, K. Pacak, N. Mullen, R. A. Wesley, and L. K. Nieman, “Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the national institutes of health,” Journal of Clinical Endocrinology & Metabolism, vol. 90, no. 8, pp. 4955–4962, 2005.View at: Publisher Site | Google Scholar
- J. Newell-Price, P. Trainer, M. Besser, and A. Grossman, “The diagnosis and differential diagnosis of cushing’s syndrome and pseudo-cushing’s states,” Endocrine Reviews, vol. 19, no. 5, pp. 647–672, 1998.View at: Publisher Site | Google Scholar
- J. Young, M. Haissaguerre, O. Viera-Pinto, O. Chabre, E. Baudin, and A. Tabarin, “Management of endocrine disease: cushing’s syndrome due to ectopic ACTH secretion: an expert operational opinion,” European Journal of Endocrinology, vol. 182, no. 4, pp. R29–R58, 2020.View at: Publisher Site | Google Scholar
- M. S. Elston, V. B. Crawford, M. Swarbrick, M. S. Dray, M. Head, and J. V. Conaglen, “Severe Cushing’s syndrome due to small cell prostate carcinoma: a case and review of literature,” Endocrine Connections, vol. 6, no. 5, pp. R80–R86, 2017.View at: Publisher Site | Google Scholar
- O. M. Alshaikh, A. A. Al-Mahfouz, H. Al-Hindi, A. B. Mahfouz, and A. S. Alzahrani, “Unusual cause of ectopic secretion of adrenocorticotropic hormone: cushing syndrome attributable to small cell prostate cancer,” Endocrine Practice, vol. 16, no. 2, pp. 249–254, 2010.View at: Publisher Site | Google Scholar
- A. Sundin, R. Arnold, E. Baudin et al., “ENETS consensus guidelines for the standards of care in neuroendocrine tumors: radiological, nuclear medicine and hybrid imaging,” Neuroendocrinology, vol. 105, no. 3, pp. 212–244, 2017.View at: Publisher Site | Google Scholar
- J.-B. Corcuff, J. Young, P. Masquefa-Giraud, P. Chanson, E. Baudin, and A. Tabarin, “Rapid control of severe neoplastic hypercortisolism with metyrapone and ketoconazole,” European Journal of Endocrinology, vol. 172, no. 4, pp. 473–481, 2015.View at: Publisher Site | Google Scholar
- L. K. Nieman, B. M. K. Biller, J. W. Findling et al., “Treatment of cushing's syndrome: an endocrine society clinical practice guideline,” Journal of Clinical Endocrinology & Metabolism, vol. 100, no. 8, pp. 2807–2831, 2015.View at: Publisher Site | Google Scholar
- T. B. Carroll, W. J. Peppard, D. J. Herrmann et al., “Continuous etomidate infusion for the management of severe cushing syndrome: validation of a standard protocol,” Journal of the Endocrine Society, vol. 3, no. 1, pp. 1–12, 2019.View at: Publisher Site | Google Scholar
- K. Von Werder, O. A. Muller, and G. K. Stalla, “Somatostatin analogs in ectopic corticotropin production,” Metabolism, vol. 45, pp. 129–131, 1996.View at: Publisher Site | Google Scholar
- N. Klomjit, D. J. Rowan, A. G. Kattah, I. Bancos, and S. J. Taler, “New-onset resistant hypertension in a newly diagnosed prostate cancer patient,” American Journal of Hypertension, vol. 32, no. 12, pp. 1214–1217, 2019.View at: Publisher Site | Google Scholar
- R. Nadal, M. Schweizer, O. N. Kryvenko, J. I. Epstein, and M. A. Eisenberger, “Small cell carcinoma of the prostate,” Nature Reviews Urology, vol. 11, no. 4, pp. 213–219, 2014.View at: Publisher Site | Google Scholar
- F. A. Collichio, P. D. Woolf, and M. Brower, “Management of patients with small cell carcinoma and the syndrome of ectopic corticotropin secretion,” Cancer, vol. 73, no. 5, pp. 1361–1367, 1994.View at: Google Scholar
Copyright
Copyright © 2022 Wanling Zeng and Joan Khoo. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
-
People sometimes ask me how I found out I had Cushing’s Disease. Theoretically, it was easy. In practice, it was very difficult.

In 1983 I came across a little article in the Ladies Home Journal which said: “If you have these symptoms…”
I found the row with my symptoms and the answer read “…ask your doctor about Cushing’s”.
After that article, I started reading everything I could on Cushing’s, I bought books that mentioned Cushing’s. I asked and asked my doctors for many years and all of them said that I couldn’t have it. It was too rare. I was rejected each time.
Due to all my reading at the library, I was sure I had Cushing’s but no one would believe me. My doctors would say that Cushing’s Disease is too rare, that I was making this up and that I couldn’t have it.
 In med school, student doctors are told “When you hear hoofbeats, think horses, not zebras“.
In med school, student doctors are told “When you hear hoofbeats, think horses, not zebras“.
According to Wikipedia: “Zebra is a medical slang term for a surprising diagnosis. Although rare diseases are, in general, surprising when they are encountered, other diseases can be surprising in a particular person and time, and so “zebra” is the broader concept.
The term derives from the aphorism “When you hear hoofbeats behind you, don’t expect to see a zebra”, which was coined in a slightly modified form in the late 1940s by Dr. Theodore Woodward, a former professor at the University of Maryland School of Medicine in Baltimore. Since horses are the most commonly encountered hoofed animal and zebras are very rare, logically you could confidently guess that the animal making the hoofbeats is probably a horse. By 1960, the aphorism was widely known in medical circles.”
So, doctors typically go for the easily diagnosed, common diseases. Just because something is rare doesn’t mean that no one gets it. We shouldn’t be dismissed because we’re too hard to diagnose.
When I was finally diagnosed in 1987, 4 years later, it was only because I started bleeding under the skin. My husband made circles around the outside perimeter each hour with a marker so my leg looked like a cut log with rings.
When I went to my Internist the next day he was shocked at the size of the rings. He now thought I had a blood disorder so he sent me to a Hematologist/Oncologist.
Fortunately, that new doctor ran a twenty-four-hour urine test and really looked at me and listened to me. Both he and his partner recognized that I had Cushing’s but, of course, couldn’t do anything further with me. They packed me off to an endo where the process started again.
My final diagnosis was in October 1987. Quite a long time to simply “…ask your doctor about Cushing’s”.
Looking back, I can see Cushing’s symptoms much earlier than 1983. But, that ‘s for a different post.

-
1
-
-

Over the years, we went on several Windjammer Barefoot Cruises. We liked them because they were small, casual and were fairly easy on the wallet.
They sailed around the Caribbean to a variety of islands, although they sometimes changed itineraries depending on weather, crew, whatever. One trip we were supposed to go to Saba but couldn’t make port. A lot of people got off at the next port and flew home.
The captains were prone to “Bedtime Stories” which were often more fiction than true but they added to the appeal of the trip. We didn’t care if we missed islands or not – we were just there to sail over the waves and enjoy the ride.
The last trip we took with them was about two years before I started having Cushing’s problems. (You wondered how I was going to tie this together, right?)
The cruise was uneventful, other than the usual mishaps like hitting docks, missing islands, and so on. Until it was a particularly rough sea one day. I was walking somewhere on deck and suddenly a wave came up over the deck making it very slippery. I fell and cracked the back of my head on the curved edge of a table in the dining area. I had the next-to-the-worse headache I have ever had, the worst being after my pituitary surgery. At least after the surgery, I got some morphine.
We asked several doctors later if that hit could have contributed to my Cushing’s but doctors didn’t want to get involved in that at all.
The Windjammer folks didn’t fare much better, either. In October 1998, Hurricane Mitch was responsible for the loss of the s/v Fantome (the last one we were on).
All 31 crew members aboard perished; passengers and other crew members had earlier been offloaded in Belize.
The story was recorded in the book The Ship and the Storm: Hurricane Mitch and the Loss of the Fantome by Jim Carrier. The ship, which was sailing in the center of the hurricane, experienced up to 50-foot (15 m) waves and over 100 mph (160 km/h) winds, causing the Fantome to founder off the coast of Honduras.
This event was similar to the Perfect Storm in that the weather people were more interested in watching the hurricane change directions than they were in people who were dealing with its effects.
I read this book and I was really moved by the plight of those crew members.
I’ll never know if that hit on my head contributed to my Cushing’s but I have seen several people mention on the message boards that they had a traumatic head injury of some type in their earlier lives.

-
She experienced extreme weight gain, thin skin and a racing heart. It took years to finally solve the medical mystery.
 Angela Yawn went to a dozen doctors before finally getting a diagnosis for her life-disrupting symptoms.Courtesy Angela Yawn
/ Source: TODAYBy A. Pawlowski
Angela Yawn went to a dozen doctors before finally getting a diagnosis for her life-disrupting symptoms.Courtesy Angela Yawn
/ Source: TODAYBy A. PawlowskiWhen a swarm of seemingly unrelated symptoms disrupted Angela Yawn’s life, she thought she was going crazy.
She gained weight — 115 pounds over six years — even as she tried to eat less. Her skin tore easily and bruises would stay on her body for months. Her face would suddenly turn blood red and hot to the touch as if she had a severe sunburn. She suffered from joint swelling and headaches. She felt tired, anxious and depressed. Her hair was falling out.
Then, there was the racing heart.
“I would put my hand on my chest because it made me feel like that’s what I needed to do to hold my heart in,” Yawn, 49, who lives in Griffin, Georgia, told TODAY.
“I noticed it during the day, but at night when I was trying to lie down and sleep, it was worse because I could do nothing but hear it beat, feel it thump."
 Yawn, seen here before the symptoms began, had no problems with weight before.Courtesy Angela Yawn
Yawn, seen here before the symptoms began, had no problems with weight before.Courtesy Angela Yawn
Yawn was especially frustrated by the weight gain. Even when she ate just 600 calories a day — consuming mostly lettuce leaves — she was still gaining about 2 pounds a day, she recalled. A doctor told her to exercise more.
 Yawn gained 115 pounds over six years. "When the weight really started to pile on, I stayed away from cameras as I felt horrible about myself and looking back at this picture is still very embarrassing for me but I wanted (people) to see what this disease has the potential to do if not diagnosed," she said.Courtesy Angela Yawn
Yawn gained 115 pounds over six years. "When the weight really started to pile on, I stayed away from cameras as I felt horrible about myself and looking back at this picture is still very embarrassing for me but I wanted (people) to see what this disease has the potential to do if not diagnosed," she said.Courtesy Angela Yawn
In all, Yawn went to a dozen doctors and was treated for high blood pressure and congestive heart failure, but nothing helped. As a last resort, she sought out an endocrinologist in February of 2021 and broke down in her office.
“That was the last hope I had of just not lying down and dying because at that point, that’s what I wanted to do,” Yawn said.
“I thought the problem was me. I thought that I’m making up these issues, that maybe I’m bipolar. I was going crazy.”
What is Cushing disease?
When the endocrinologist suddenly started listing all of her symptoms without being prompted, Yawn stopped crying.
Blood tests and an MRI finally confirmed the doctor’s suspicion: Yawn had a tumor in her pituitary gland — a pea-size organ at the base of the brain — that was causing the gland to release too much adrenocorticotropic hormone. That, in turn, flooded her body with cortisol, a steroid hormone that’s normally released in response to stress or danger. The resulting condition is called Cushing disease.
Imagine the adrenaline rush you’d get while jumping out of an airplane and skydiving — that’s what Yawn felt all the time, with harmful side-effects.
Yawn was making six times the cortisol she needed, said Dr. Nelson Oyesiku, chair of neurosurgery at UNC Health in Chapel Hill, North Carolina, who removed her tumor last fall.
“That’s a trailer load of cortisol. Day in, day out, morning, noon and night, whether you need it or not, your body just keeps making this excess cortisol. It can wreak havoc in the body physiology and metabolism,” Oyesiku told TODAY.
The steroid regulates blood pressure and heart rate, which is why Yawn's skin was flushed and her heart was racing, he noted. It can regulate how fat is burned and deposited in the body, which is why Yawn was gaining weight. Other effects of the steroid's overproduction include fatigue, thin skin with easy bruising, mental changes and high blood sugar.
Cushing disease is rare, affecting about five people per million each year, so most doctors will spend their careers without ever coming across a case, Oyesiku said. That’s why patients often go years without being diagnosed: When they complain of blood sugar problems or a racing heart, they’ll be treated for much more common issues like diabetes or high blood pressure.
Pituitary gland is hard to reach
Removing Yawn’s tumor in September of 2021 would require careful maneuvering.
If you think of the head as a ball, the pituitary gland sits right at the center, between the ears, between the eyes and about 4 inches behind the nose, Oyesiku said. It’s called the “master gland” because it regulates other glands in the body that make hormones, he noted.
 The location of the pituitary gland makes it heard to reach.janulla / Getty Images
The location of the pituitary gland makes it heard to reach.janulla / Getty Images
It’s a very difficult spot to reach. To get to it, Oyesiku made an incision deep inside Yawn’s nose in a small cavity called the sphenoid sinus. Using a long, thin tube that carried a light and a camera, he reached the tiny tumor — about the size of a rice grain — and removed it using special instruments. The surgery took four hours.
The potential risk is high: The area is surrounded by vessels that carry blood to the brain, and it’s right underneath optic nerves necessary for a person to see. If things go wrong, patients can become blind, brain dead, or die.
Recovery from surgery
Today, Yawn is slowly returning to normal. She has lost 41 pounds and continues to lose weight. Her hair is no longer falling out.
But patients sometimes require months or even a few years to adjust to normal cortisol levels.
“It takes some time to unwind the effects of chronic exposure to steroids, so your body has to adapt to the new world order as the effects of the steroids recede,” Oyesiku said.
 "My life was on hold for five years... I'm trying not to be too impatient," Yawn said.Courtesy Angela Yawn
"My life was on hold for five years... I'm trying not to be too impatient," Yawn said.Courtesy Angela Yawn
Yawn’s body was so used to that higher cortisol level that she’s had to rely on steroid supplements to feel normal after the surgery. It’s like an addict going through withdrawal, she noted.
The next step is finishing another cycle of supplements and then slowly tapering off them so that her body figures out how to function without the steroid overload.
“I am definitely moving in the right direction,” she said. "I hope that I’ll get back to that woman I used to be — in mind, body and spirit."
-
1
-
-
I don't know if there's anything of interest here - or the cost - but possibly useful to someone.
Cushing’s Syndrome Diagnostic and Treatment Market research report is the new statistical data source added by Research Cognizance.
“Cushing’s Syndrome Diagnostic and Treatment Market is growing at a High CAGR during the forecast period 2022-2029. The increasing interest of the individuals in this industry is that the major reason for the expansion of this market”.
Cushing’s Syndrome Diagnostic and Treatment Market research is an intelligence report with meticulous efforts undertaken to study the right and valuable information. The data which has been looked upon is done considering both, the existing top players and the upcoming competitors. Business strategies of the key players and the new entering market industries are studied in detail. Well explained SWOT analysis, revenue share, and contact information are shared in this report analysis.
Get the PDF Sample Copy (Including FULL TOC, Graphs, and Tables) of this report @:
https://researchcognizance.com/sample-request/896
Top Key Players Profiled in this report are:
Novartis, Orphagen Pharmaceuticals, Inc., Corcept Therapeutics
The key questions answered in this report:
- What will be the Market Size and Growth Rate in the forecast year?
- What are the Key Factors driving Cushing’s Syndrome Diagnostic and Treatment Market?
- What are the Risks and Challenges in front of the market?
- Who are the Key Vendors in Cushing’s Syndrome Diagnostic and Treatment Market?
- What are the Trending Factors influencing the market shares?
- What are the Key Outcomes of Porter’s five forces model?
- Which are the Global Opportunities for Expanding the Cushing’s Syndrome Diagnostic and Treatment Market?
Various factors are responsible for the market’s growth trajectory, which are studied at length in the report. In addition, the report lists down the restraints that are posing threat to the global Cushing’s Syndrome Diagnostic and Treatment market. It also gauges the bargaining power of suppliers and buyers, threat from new entrants and product substitute, and the degree of competition prevailing in the market. The influence of the latest government guidelines is also analyzed in detail in the report. It studies the Cushing’s Syndrome Diagnostic and Treatment market’s trajectory between forecast periods.
Get up to 30% Discount on this Premium Report @:
https://researchcognizance.com/discount/896
Regions Covered in the Global Cushing’s Syndrome Diagnostic and Treatment Market Report 2022:
• The Middle East and Africa (GCC Countries and Egypt)
• North America (the United States, Mexico, and Canada)
• South America (Brazil etc.)
• Europe (Turkey, Germany, Russia UK, Italy, France, etc.)
• Asia-Pacific (Vietnam, China, Malaysia, Japan, Philippines, Korea, Thailand, India, Indonesia, and Australia)The cost analysis of the Global Cushing’s Syndrome Diagnostic and Treatment Market has been performed while keeping in view manufacturing expenses, labor cost, and raw materials and their market concentration rate, suppliers, and price trend. Other factors such as Supply chain, downstream buyers, and sourcing strategy have been assessed to provide a complete and in-depth view of the market. Buyers of the report will also be exposed to a study on market positioning with factors such as target client, brand strategy, and price strategy taken into consideration.
The report provides insights on the following pointers:
Market Penetration: Comprehensive information on the product portfolios of the top players in the Cushing’s Syndrome Diagnostic and Treatment market.
Product Development/Innovation: Detailed insights on the upcoming technologies, R&D activities, and product launches in the market.
Competitive Assessment: In-depth assessment of the market strategies, geographic and business segments of the leading players in the market.
Market Development: Comprehensive information about emerging markets. This report analyzes the market for various segments across geographies.
Market Diversification: Exhaustive information about new products, untapped geographies, recent developments, and investments in the Cushing’s Syndrome Diagnostic and Treatment market.
Table of Content Global Cushing’s Syndrome Diagnostic and Treatment Market Research Report Chapter 1: Global Cushing’s Syndrome Diagnostic and Treatment Industry Overview Chapter 2: Global Economic Impact on Cushing’s Syndrome Diagnostic and Treatment Industry Chapter 3: Global Market Competition by Industry Producers Chapter 4: Global Productions, Revenue (Value), according to regions Chapter 5: Global Supplies (Production), Consumption, Export, Import, geographically Chapter 6: Global Productions, Revenue (Value), Price Trend, Product Type Chapter 7: Global Market Analysis, on the basis of Application Chapter 8: Cushing’s Syndrome Diagnostic and Treatment Market Pricing Analysis Chapter 9: Market Chain, Sourcing Strategy, and Downstream Buyers Chapter 10: Strategies and key policies by Distributors/Suppliers/Traders Chapter 11: Key Marketing Strategy Analysis, by Market Vendors Chapter 12: Market Effect Factors Analysis Chapter 13: Global Cushing’s Syndrome Diagnostic and Treatment Market Forecast Buy Exclusive Report @:
https://researchcognizance.com/checkout/896/single_user_license
If you have any special requirements, please let us know and we will offer you the report as you want.
About Us:
Research Cognizance is an India-based market research Company, registered in Pune. Research Cognizance aims to provide meticulously researched insights into the market. We offer high-quality consulting services to our clients and help them understand prevailing market opportunities. Our database presents ample statistics and thoroughly analyzed explanations at an affordable price.
Contact Us:
Neil Thomas
116 West 23rd Street 4th Floor New York City, New York 10011
+1 7187154714
-
1
-
We have an opportunity for you to take part in a Acromegaly and Human Growth Hormone Deficiency (GHD) Study for patients and caregivers. Our project number for this study is IQV_6382.
Project Details:
- Survey is 20-minutes long
- $35 Reward
Things to Note:
- We recommend using the web browsers Google Chrome or FireFox
- Study is open to patients and caregivers
- Please do not share study links
- One participant per household only
- Want to share this opportunity? Let us know and we can provide a new link
- Please use a laptop/computer ONLY. No smartphones or tablets - Preliminary questions are mobile friendly!
- Save this email to reference if you have any questions about the study!
-
If you have any problems, email lejla.zonic@
rarepatientvoice.com and reference the project number.
If you are interested in this study, please sign up for Rare Patient Voice here: https://rarepatientvoice.com/
CushingsHelp/ Thanks as always for your participation! Please be aware that by entering this information you are not guaranteed that you will be selected to participate. As always, we do not share any of your contact information without your permission.
-
1
-
Under a Creative Commons licenseOpen access
Highlights
- • We describe a rare case of a patient with a sparsely granulated corticotroph pituitary macroadenoma with pituitary apoplexy who underwent transsphenoidal resection resulting in remission of hypercortisolism.
- • Corticotroph adenomas are divided into densely granulated, sparsely granulated and Crooke’s cell tumors.
- • macroadenomas account for 7-23% of patients with pituitary corticotroph adenomas
- • Sparsely granulated corticotroph tumors are associated with longer duration of Cushing disease prior to diagnosis, larger tumor size at diagnosis, decreased immediate remission rate, increased proliferative marker Ki-67 and increased recovery time of hypothalamic-pituitary-adrenal axis after surgery.
- • Granulation pattern is an important clinicopathological distinction impacting the behavior and treatment outcomes of pituitary corticotroph adenomas
Abstract
Background
/Objective: Pituitary corticotroph macroadenomas, which account for 7% to 23% of corticotroph adenomas, rarely present with apoplexy. The objective of this report is to describe a patient with a sparsely granulated corticotroph tumor (SGCT) presenting with apoplexy and remission of hypercortisolism.
Case Report
A 33-year-old male presented via ambulance with sudden onset of severe headache and nausea/vomiting. Physical exam revealed bitemporal hemianopsia, diplopia from right-sided third cranial nerve palsy, abdominal striae, facial plethora, dorsal and supraclavicular fat pad. Magnetic resonance imaging (MRI) demonstrated a 3.2 cm mass arising from the sella turcica with hemorrhage compressing the optic chiasm, extension into the sphenoid sinus and cavernous sinus. Initial investigations revealed plasma cortisol of 64.08 mcg/dL (Reference Range (RR), 2.36 – 17.05). He underwent emergent transsphenoidal surgery. Pathology was diagnostic of SGCT. Post-operatively, cortisol was <1.8ug/dL (RR, 2.4 – 17), adrenocorticotropic hormone (ACTH) 36 pg/mL (RR, 0 – 81), thyroid stimulating hormone (TSH) 0.07 uIU/mL (RR, 0.36 - 3.74), free thyroxine 1 ng/dL (RR, 0.8 – 1.5), luteinizing hormone (LH) <1 mIU/mL (RR, 1 – 12), follicle stimulating hormone (FSH) 1 mIU/mL (RR, 1 – 12) and testosterone 28.8 ng/dL (RR, 219.2 – 905.6) with ongoing requirement for hydrocortisone, levothyroxine, testosterone replacement and continued follow-up.
Discussion
Corticotroph adenomas are divided into densely granulated, sparsely granulated and Crooke’s cell tumors. Sparsely granulated pattern is associated with larger tumor size and decreased remission rate after surgery.
Conclusion
This report illustrates a rare case of hypercortisolism remission due to apoplexy of a SGCT with subsequent central adrenal insufficiency, hypothyroidism and hypogonadism.
Keywords
pituitary apoplexypituitary macroadenomapituitary tumorsparsely granulated corticotroph tumorCushing diseaseIntroduction
The incidence of Cushing Disease (CD) is estimated to be between 0.12 to 0.24 cases per 100,00 persons per year1,2. Of these, 7-23% are macroadenomas (>1 cm)3, 4, 5. Pituitary apoplexy is a potentially life-threatening endocrine and neurosurgical emergency which occurs due to infarction or hemorrhage in the pituitary gland. Apoplexy occurs most commonly in non-functioning macroadenomas with an estimated prevalence of 6.2 cases per 100,000 persons and incidence of 0.17 cases per 100,00 persons per year6. Corticotroph macroadenoma presenting with apoplexy is uncommon with only a handful of reports in the literature7. We present a case of a sparsely granulated corticotroph (SGCT) which presented with apoplexy leading to remission of hypercortisolism and subsequent central adrenal insufficiency.
Case Presentation
A 33-year-old male who was otherwise healthy and not on any medications presented to a community hospital with sudden and severe headache accompanied by hypotension, nausea, vomiting, bitemporal hemianopsia and diplopia. Computed Tomography (CT) scan of the brain demonstrated a hyperattenuating 2.0 cm x 2.8 cm x 1.5 cm mass at the sella turcica with extension into the right cavernous sinus and encasement of the right internal carotid arteries (Figure 1A). He was transferred to a tertiary care center for neurosurgical management with endocrinology consultation post-operatively.

Figure 1. hyperattenuating 2.0 cm x 2.8 cm x 1.5 cm mass at the sella turcica on unenhanced CT (A); MRI demonstrated a 1.9 cm x 3.2 cm x 2.4 cm heterogeneous mass on T1 (B) and T2-weighted imaging (C) showing small hyperintense areas in solid part of the sella mass with flattening of the optic chiasm, remodeling/dehiscence of the floor of the sella and extending into the right cavernous sinus with at least partial encasement of the ICA
In retrospect, he reported a 3-year history of ongoing symptoms of hypercortisolism including increased central obesity, dorsal and supraclavicular fat pad, facial plethora, abdominal purple striae, easy bruising, fatigue, decreased libido and erectile dysfunction. Notably, at the time of presentation he did not have a history of diabetes, hypertension, osteoporosis, fragility fractures or proximal muscle weakness. He fathered 2 children previously. His physical examination was significant for Cushingoid facies, facial plethora, dorsal and supraclavicular fat pads and central obesity with significant axillary and abdominal wide purple striae (Figure 2). Neurological examination revealed bitemporal hemianopsia, right third cranial nerve palsy with ptosis and impaired extraocular movement. The fourth and sixth cranial nerves were intact as was the rest of his neurological exam. These findings were corroborated by Ophthalmology.

Figure 2. Representative images illustrating facial plethora (A); abdominal striae (B, C); supraclavicular fat pad (D); dorsal fat pad (E)
Initial laboratory data at time of presentation to the hospital included elevated plasma cortisol of 64.08ug/dL (RR, 2.36 – 17.05), ACTH was not drawn at the time of presentation, normal TSH 0.89 mIU/L (RR, 0.36 – 3.74), free thyroxine 0.91ng/dL (RR, 0.76 – 1.46), evidence of central hypogonadism with low total testosterone 28.8 ng/dL (RR, 219.2 – 905.6) and inappropriately normal luteinizing hormone (LH) 1mIU/mL (RR, 1 – 12) and follicle stimulating hormone (FSH) 3mIU/mL (RR, 1 – 12), low prolactin <1 ng/mL (RR, 3 – 20), and normal insulin growth factor – 1 (IGF–1) 179ng/mL (RR, 82 – 242).
A pituitary gland dedicated MRI was performed to further characterize the mass, which re-demonstrated a 1.9 cm x 3.2 cm x 2.4 cm heterogenous mass at the sella turcica extending superiorly and flattening the optic chiasm, remodeling of the floor of the sella and bulging into the sphenoid sinus and extending laterally into the cavernous sinus with encasement of the right internal carotid artery (ICA). As per the radiologist’s diagnostic impression, this appearance was most in keeping with a pituitary macroadenoma with apoplexy (Figure 1B – C).
The patient underwent urgent TSS and decompression with no acute complications. Pathological examination of the pituitary adenoma showed features characteristic of sparsely granulated corticotroph pituitary neuroendocrine tumor (adenoma)8, with regional hemorrhage and tumor necrosis (apoplexy). The viable tumor exhibited a solid growth pattern (Figure 3A), t-box transcription factor (T-pit) nuclear immunolabeling (Figure 3B), diffuse cytoplasmic CAM5.2 (low molecular weight cytokeratin) immunolabeling (Figure 3C), and regional weak to moderate intense granular cytoplasmic ACTH immuno-staining (Figure 3D). The tumor was immuno-negative for: pituitary-specific positive transcription factor 1 (Pit-1) and steroidogenic factor 1 (SF-1) transcription factors, growth hormone, prolactin, TSH, FSH, LH, estrogen receptor-alpha, and alpha-subunit. Crooke hyalinization was not identified in an adjacent compressed fragment of non-adenomatous anterior pituitary tissue. Ki-67 immunolabeling showed a 1.5% proliferative index (11 of 726 nuclei).

Figure 3. Hematoxylin phloxine saffron staining showing adenoma with solid growth pattern (A); immunohistochemical staining showing T-pit reactivity of tumor nuclei (B); diffuse cytoplasmic staining for cytokeratin CAM5.2 (C); and regional moderately intense granular cytoplasmic staining for ACTH (D). Scale bar = 20 μm
Post-operatively, he developed transient central diabetes insipidus requiring desmopressin but resolved on discharge. His postoperative cortisol was undetectable, ACTH 36 pg/mL (RR, 0 - 81), TSH 0.07 mIU/mL (RR, 0.36 - 3.74), free thyroxine 1 ng/dL (RR, 0.8 - 1.5), LH <1mIU/mL (RR, 1 - 12), FSH 1 mIU/mL (RR, 1 - 12) and testosterone 28.8 ng/dL (RR, 219.2 - 905.6) (Table 1 and Figure 4). One month later, he reported 15 pounds of weight loss and a 5-inch decrease in waist circumference. He also noted a reduction in the dorsal and supraclavicular fat pads, facial plethora, and Cushingoid facies as well as fading of the abdominal stretch marks. His visual field defects and right third cranial nerve palsy resolved on follow up with ophthalmology post-operatively. Repeat MRI six months post-operatively showed minor residual soft tissue along the floor of the sella. He is being followed by Neurosurgery, Ophthalmology, and Endocrinology for monitoring of disease recurrence, visual defects, and management of hypopituitarism.
Table 1. Pre- and post-operative hormonal panel
POD -1 POD 0 POD1 POD2 POD3 POD16 6 -9 months Comments Cortisol(2.4 – 17 ug/dL) 64↓ 32↓ 11↓ <1.8↓ <1.8↓ 1.8↓ HC started POD3 post bloodwork ACTH(0 – 81 pg/mL) 41↓ 36↓ 28↓ 13↓ TSH(0.36 - 3.74 uIU/mL) 0.89 0.43 0.12↓ 0.07↓ 0.05↓ 0.73 Thyroxine, free(0.8 – 1.5 ng/dL) 0.9 0.9 1.1 1 2.1↑ 1 Levothyroxine started POD4 LH(1 – 12 miU/mL) 1↓ <1↓ 1↓ 3 FSH(1 – 12 mIU/mL) 3↓ 1↓ 1↓ 3 Testosterone(219.2 – 905.6 ng/dL) 28.8↓ <20↓ 175.9↓ Testosterone replacement started as outpatient Testosterone, free(160 - 699 pmol/L) <5.8↓ 137↓ IGF-1(82 – 242 ng/mL) 179 79 GH(fasting < 6 mIU/L) 4.5 <0.3 Prolactin(3 – 20 ng/mL) <1↓ <1↓ POD, postoperative day; HC, hydrocortisone; ACTH, adrenocorticotropic hormone; TSH, thyroid stimulating hormone; LH, luteinizing Hormone; FSH, follicle stimulating hormone; IGF-1, insulin like growth factor - 1; GH, growth hormone

Figure 4. Trend of select pituitary hormonal panel with key clinical events denoted by black arrows.
Discussion
Microadenomas account for the majority of corticotroph tumors, but 7% – 23% of patients are diagnosed with a macroadenoma3, 4, 5. It is even rarer for a corticotroph macroadenoma to present with apoplexy with only a handful of case reports or series in the literature7. Due to its rarity, appropriate biochemical workup on presentation, such as including an ACTH with the blood work, may be omitted especially if the patient is going for emergent surgery. In this case, the undetectable prolactin can reflect loss of anterior pituitary function and also suggest a functioning corticotroph adenoma due to the inhibitory effect of long term serum glucocorticoids on prolactin secretion9. After undergoing TSS, the patient developed central adrenal insufficiency, hypothyroidism and hypogonadism requiring hormone replacement. Presumably, the development of adrenal insufficiency demonstrated the remission of hypercortisolism as a result of apoplexy and/or TSS. The ACTH remains detectable likely representing residual tumor that was not obliterated by apoplexy nor excised by TSS given it location near the carotid artery and cavernous sinus. The presence of adrenal insufficiency in the setting of detectable ACTH is not contradictory as the physiological hypothalamic-pituitary-adrenal axis has been suppressed by the long-term pathological production of ACTH. IGF-1 and prolactin also failed to recover post-operatively. In CD where the production of IGF-1 and prolactin are attenuated by elevated cortisol, it would then be expected that IGF-1 and prolactin recover after hypercortisolism remission. However, the absence of this observation in our case is likely a sequalae of the apoplexy and extensive surgery leading to pituitary hypofunction.
We also want to highlight features of the pre-operative radiographical findings which can provide valuable insight into the subsequent histology. Previous literature has shown that, on T2-weight MRI, silent corticotroph adenomas are strongly correlated with characteristic a multimicrocystic appearance while nonfunctional gonadotroph macroadenomas are not correlated with this MRI finding10. The multimicrocystic appearance is described as small hyperintense areas with hyperintense striae in the solid part of the tumor (Figure 1C)10. This is an useful predictive tool for silent corticotroph adenomas with a sensitivity of 76%, specificity of 95% and a likelihood ratio of 15.310.
The ability to distinguish between silent corticotroph macroadenoma and other macroadenomas is important for assessing rate of remission and recurrence risk. In 2017, the WHO published updated classification for pituitary tumors. In this new classification, corticotroph adenomas are further divided into densely granulated, sparsely granulated and Crooke’s cell tumors11. DGCT are intensely Periodic Acid Schiff (PAS) stain positive and exhibit strong diffuse pattern of ACTH immunoreactivity, whereas SGCT exhibit faintly positive PAS alongside weak focal ACTH immunoreactivity4,12. Crooke’s cell tumors are characterized by Crooke’s hyaline changes in more than 50% of the tumor cells4. In the literature, SGCT account for an estimated 19-29% of corticotroph adenomas13, 14, 15. The clinicopathological relevance of granulation pattern in corticotroph tumors was unclear until recently.
In multiple studies examining granulation pattern and tumor size, SGCT were statistically larger13,15,16. Hence, we suspect that many of the previously labelled silent corticotroph macroadenomas in the literature were SGCT. The traditional teaching of CD has been “small tumor, big Cushing and big tumor, small Cushing” which reflects the inverse relationship between tumor size and symptomatology17. This observation appears to hold true as Doğanşen et al. found a trend towards longer duration of CD in SGCT of 34 months compared to 26 months in DGCT based on patient history13,17. It has been postulated that the underlying mechanism of the inverse relationship between tumor size and symptomatology is impaired processing of proopiomelanocortin resulting in less effective secretion of ACTH in corticotroph macroadenomas3. Doğanşen et al. also found that the recurrence rate was doubled for SGCT, while Witek et al. showed that SGCT were less likely to achieve remission postoperatively13,16.
Similar to other cases of SGCT, the diagnosis was only arrived retrospective after pathological confirmation10. Interestingly, the characteristic Crooke’s hyaline change of surrounding non-adenomatous pituitary tissue was not observed as one would expect in a state of prolonged glucocorticoid excess in this case. Although classically described, the absence of this finding does not rule out CD. As evident in a recent retrospective study where 10 out of 144 patients with CD did not have Crooke’s hyaline change18. In patients without Crooke’s hyaline change, the authors found a lower remission rate of 44.4% compared to 73.5% in patients with Crooke’s hyaline change. Together with the detectable post-operative ACTH, sparsely granulated pattern and absence of Crooke’s hyaline change in surrounding pituitary tissue, the risk of recurrence is increased. These risk factors emphasize the importance of close monitoring to ensure early detection of recurrence.
Declaration of Interests
☒ The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
☐The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Conclusion
We present a case of a sparsely granulated corticotroph macroadenoma presenting with apoplexy leading to remission of hypercortisolism and development of central adrenal insufficiency, hypothyroidism and hypogonadism requiring hormone replacement.
References
- 1
-
J. Lindholm, S. Juul, J.O. Jørgensen, et al.Incidence and late prognosis of cushing's syndrome: a population-based studyJ Clin Endocrinol Metab, 86 (1) (2001), pp. 117-123
- 2
-
J. Etxabe, J.A. VazquezMorbidity and mortality in Cushing's disease: an epidemiological approachClin Endocrinol (Oxf), 40 (4) (1994), pp. 479-484
- 3
-
L. Katznelson, J.S. Bogan, J.R. Trob, et al.Biochemical assessment of Cushing's disease in patients with corticotroph macroadenomasJ Clin Endocrinol Metab, 83 (5) (1998), pp. 1619-1623
- 4
-
L.V. Syro, F. Rotondo, M.D. Cusimano, et al.Current status on histological classification in Cushing's diseasePituitary, 18 (2) (2015), pp. 217-224
- 5
-
Y.S. Woo, A.M. Isidori, W.Z. Wat, et al.Clinical and biochemical characteristics of adrenocorticotropin-secreting macroadenomasJ Clin Endocrinol Metab, 90 (8) (2005), pp. 4963-4969
- 6
-
C. Briet, S. Salenave, J.F. Bonneville, E.R. Laws, P. ChansonPituitary ApoplexyEndocr Rev, 36 (6) (2015), pp. 622-645
- 7
-
K. Siwakoti, S.B. Omay, S.E. InzucchiSPONTANEOUS RESOLUTION OF PRIMARY HYPERCORTISOLISM OF CUSHING DISEASE AFTER PITUITARY HEMORRHAGEAACE Clin Case Rep, 6 (1) (2020), pp. e23-e29
- 8
-
S.L. Asa, O. MeteWhat's new in pituitary pathology?Histopathology, 72 (1) (2018), pp. 133-141
- 9
-
M.E. Freeman, B. Kanyicska, A. Lerant, G. NagyProlactin: Structure, Function, and Regulation of SecretionPhysiological Reviews, 80 (4) (2000), pp. 1523-1631
- 10
-
L. Cazabat, M. Dupuy, A. Boulin, et al.Silent, but not unseen: multimicrocystic aspect on T2-weighted MRI in silent corticotroph adenomasClin Endocrinol (Oxf), 81 (4) (2014), pp. 566-572
- 11
-
M.B.S. LopesThe 2017 World Health Organization classification of tumors of the pituitary gland: a summaryActa Neuropathol, 134 (4) (2017), pp. 521-535
- 12
-
W. Saeger, J. Honegger, M. Theodoropoulou, et al.Clinical Impact of the Current WHO Classification of Pituitary AdenomasEndocr Pathol, 27 (2) (2016), pp. 104-114
- 13
-
S. Doğanşen, B. Bilgiç, G.Y. Yalin, S. Tanrikulu, S. YarmanClinical Significance of Granulation Pattern in Corticotroph Pituitary AdenomasTurk Patoloji Derg, 35 (1) (2019), pp. 9-14
- 14
-
O. Mete, A. Cintosun, I. Pressman, S.L. AsaEpidemiology and biomarker profile of pituitary adenohypophysial tumorsMod Pathol, 31 (6) (2018), pp. 900-909
- 15
-
B. Rak, M. Maksymowicz, M. Pękul, G. ZielińskiClinical, Biological, Radiological Pathological and Immediate Post-Operative Remission of Sparsely and Densely Granulated Corticotroph Pituitary Tumors: A Retrospective Study of a Cohort of 277 Patients With Cushing's DiseaseFront Endocrinol (Lausanne), 12 (2021)672178
- 16
-
P. Witek, G. Zieliński, K. Szamotulska, M. Maksymowicz, G. KamińskiClinicopathological predictive factors in the early remission of corticotroph pituitary macroadenomas in a tertiary referral centreEur J Endocrinol, 174 (4) (2016), pp. 539-549
- 17
-
A.M. McNicolTumors of the pituitary gland. S. L. Asa. AFIP atlas of tumor pathology, third seriesThe Journal of Pathology, 188 (1) (1999), pp. 115-116
- 18
-
A. Akirov, V. Larouche, I. Shimon, et al.Significance of Crooke's Hyaline Change in Nontumorous Corticotrophs of Patients With Cushing DiseaseFront Endocrinol (Lausanne), 12 (2021)620005
-
1
-

This is another semi-religious post so feel free to skip it 🙂
I’m sure that many would think that Abide With Me is a pretty strange choice for my all-time favorite hymn.
My dad was a Congregational (now United Church of Christ) minister so I was pretty regular in church attendance in my younger years.
Some Sunday evenings, he would preach on a circuit and I’d go with him to some of these tiny churches. The people there, mostly older folks, liked the old hymns best – Fanny Crosby and so on.
So, some of my “favorite hymns” are those that I sang when I was out with my Dad. Fond memories from long ago.
In 1986 I was finally diagnosed with Cushing’s after struggling with doctors and trying to get them to test for about 5 years. I was going to go into the NIH (National Institutes of Health) in Bethesda, MD for final testing and then-experimental pituitary surgery.
I was terrified and sure that I wouldn’t survive the surgery.
Somehow, I found a 3-cassette tape set of Readers Digest Hymns and Songs of Inspiration and ordered that. The set came just before I went to NIH and I had it with me.
At NIH I set up a daily “routine” of sorts and listening to these tapes was a very important part of my day and helped me get through the ordeal of more testing, surgery, post-op and more.
When I had my kidney cancer surgery, those tapes were long broken and irreplaceable, but I had replaced all the songs – this time on my iPod.
Abide With Me was on this original tape set and it remains a favorite to this day. Whenever we have an opportunity in church to pick a favorite, my hand always shoots up and I request page 700. When someone in one of my handbell groups moves away, we always sign a hymnbook and give it to them. I sign page 700.
I think that many people would probably think that this hymn is depressing. Maybe it is but to me it signifies times in my life when I thought I might die and I was so comforted by the sentiments here.
This hymn is often associated with funeral services and has given hope and comfort to so many over the years – me included.
If you abide in Me, and My words abide in you, you will ask what you desire, and it shall be done for you.
~John 15:7
Abide With Me
Words: Henry F. Lyte, 1847.
Music: Eventide, William H. Monk, 1861. Mrs. Monk described the setting:
This tune was written at a time of great sorrow—when together we watched, as we did daily, the glories of the setting sun. As the last golden ray faded, he took some paper and penciled that tune which has gone all over the earth.
Lyte was inspired to write this hymn as he was dying of tuberculosis; he finished it the Sunday he gave his farewell sermon in the parish he served so many years. The next day, he left for Italy to regain his health. He didn’t make it, though—he died in Nice, France, three weeks after writing these words. Here is an excerpt from his farewell sermon:
O brethren, I stand here among you today, as alive from the dead, if I may hope to impress it upon you, and induce you to prepare for that solemn hour which must come to all, by a timely acquaintance with the death of Christ.
For over a century, the bells of his church at All Saints in Lower Brixham, Devonshire, have rung out “Abide with Me” daily. The hymn was sung at the wedding of King George VI, at the wedding of his daughter, the future Queen Elizabeth II, and at the funeral of Nobel peace prize winner Mother Teresa of Calcutta in1997.
Abide with me; fast falls the eventide;
The darkness deepens; Lord with me abide.
When other helpers fail and comforts flee,
Help of the helpless, O abide with me.
Swift to its close ebbs out life’s little day;
Earth’s joys grow dim; its glories pass away;
Change and decay in all around I see;
O Thou who changest not, abide with me.
Not a brief glance I beg, a passing word;
But as Thou dwell’st with Thy disciples, Lord,
Familiar, condescending, patient, free.
Come not to sojourn, but abide with me.
Come not in terrors, as the King of kings,
But kind and good, with healing in Thy wings,
Tears for all woes, a heart for every plea—
Come, Friend of sinners, and thus bide with me.
Thou on my head in early youth didst smile;
And, though rebellious and perverse meanwhile,
Thou hast not left me, oft as I left Thee,
On to the close, O Lord, abide with me.
I need Thy presence every passing hour.
What but Thy grace can foil the tempter’s power?
Who, like Thyself, my guide and stay can be?
Through cloud and sunshine, Lord, abide with me.
I fear no foe, with Thee at hand to bless;
Ills have no weight, and tears no bitterness.
Where is death’s sting? Where, grave, thy victory?
I triumph still, if Thou abide with me.
Hold Thou Thy cross before my closing eyes;
Shine through the gloom and point me to the skies.
Heaven’s morning breaks, and earth’s vain shadows flee;
In life, in death, O Lord, abide with me.
http://cushieblog.files.wordpress.com/2012/04/maryo-butterfly-script1.gif?resize=251%2C121
-
1
-
-

I wrote parts of this in 2008, so all the “yesterdays” and “last weeks” are a little off.
Wow. That’s about all I can say. Yesterday was possibly the best day of my life since I started getting Cushing’s symptoms, and that was over 30 years ago. More than a quarter of a century of feeling exhausted, fatigued. A quarter of my life spent taking naps and sleeping.
Last week in this post I wrote in part:
I went to the endo yesterday. Nothing has changed for me. Nothing will. He wants me to take more cortef. I don’t want to gain weight again. He looked up Provigil and it’s not indicated for panhypopituitarism. So he won’t prescribe it. My kidney surgeon probably won’t let me take, anyway, but it was worth a try.
…
He did mention that in “only” 2.5 years maybe I can go back on growth hormone. I don’t want to live like this another year let alone 2.5. But then, when I was on GH before it didn’t help me like it helps most everyone else.
I’m tired of catering to a kidney that may or may not fail sometime anyway, tired of being so exhausted all the time. I feel like I’ve lost nearly half my life to this Cushing’s stuff already.
So, yesterday I was supposed to go to a conference on web design for churches. My church sent me because they want me to spiff up their site and make them a new one for Christmas. I wanted to go because, well, I like learning new stuff about the web. I figured that I would learn stuff that would also be useful to me in others of my sites.
And I did!
But the amazing thing is this. My son had told me about a medication that was very similar to Provigil, that he had tried it while he was writing his doctoral thesis and it had helped him.
So, having tried the official doctor route and being rebuffed – again – I had decided to try this stuff on my own.
Just the night before I had written a response on Robin’s wonderful blog that reads in part:
I hate this disease, too.
I was just talking to a friend today about how I’d try nearly anything – even if it ruined my one remaining kidney – to have a few days where I felt good, normal, where I could wake up in the morning rested and be able to have energy for the day.
I want to go out and have fun, to be able to drive for more than 45 minutes without needing to rest, to be have people over for dinner, whatever. I hate being restricted by my lack of energy.
My endo says to cheer up. In two and a half years I can try the growth hormone again. Whoopee. Didn’t work the first time and maybe gave me, or contributed to, cancer growth. Why would I want to look forward to trying that again?
I want to feel good now. Today.
I hate that this disease kills but I also hate that it’s robbed me of half my life already.
I wish doctors would understand that even though we’ve “survived”, there’s no quality of life there.
I hate Cushing’s. It robs so much from so many of us. 🙁
As I said earlier, I have a history of daily naps of at least 3 hours a day. It cuts into everything and prevents me from doing many things. I have to schedule my life around these naps and it’s awful.
 A few years ago I went on a Cushie trip to Rockford. I’ve been there a few times and it’s always so much fun. But this first year, we were going to another Cushie’s home for barbecue. I didn’t drive, I rested in the back of the car during the drive. We got there and I managed to stay awake for a little while. Them I put my head down on the dining room table and fell asleep. Our hostess kindly suggested that I move over to the sofa.
A few years ago I went on a Cushie trip to Rockford. I’ve been there a few times and it’s always so much fun. But this first year, we were going to another Cushie’s home for barbecue. I didn’t drive, I rested in the back of the car during the drive. We got there and I managed to stay awake for a little while. Them I put my head down on the dining room table and fell asleep. Our hostess kindly suggested that I move over to the sofa.
So, I have a long history of daily naps, not getting through the day, yadda, yadda.
So, I was a little nervous about yesterday. I really wanted to go to this conference, and was afraid I’d have to go nap in my car.
I got up at 5:30 am yesterday. Before I left at 7:15, I took my Cortef and then I took my non-FDA approved simulated Provigil. (Although it’s not FDA approved, it is not illegal to possess without a prescription and can be imported privately by citizens)
I stayed awake for the whole conference, went to a bell rehearsal, did Stacey’s interview, had dinner and went to bed about 10:30PM. NO NAP! I did close my eyes a little during the 4:00PM session but it was also b-o-r-i-n-g.
I stayed awake, I enjoyed myself, I learned stuff, I participated in conversations (completely unlike shy me!).
I felt like I think normal people feel. I was amazed. Half my life wasted and I finally (thank you Michael!) had a good day.
My kidney doctor and my endo would probably be appalled but it’s about time that I had some life again! Maybe in another 25 years, I’ll take another pill. LOL
Well, the energy from the Adrafinil was a one day thing. I felt great on Thursday. Friday and Saturday I slept more than usual. Saturday, today, was one of those days where I sleep nearly all day. Maybe if I took the drug more it would build up in my system, maybe not. But it was still worth having that one day where I felt what I imagine normal to be.
While I was being a slug today, my husband painted the entire house.
I’m not sure if I would have been this tired today or if I was somehow making up for the nap I didn’t get on Thursday. Whatever the case, I’m glad that I had the opportunity to try this and to experience the wonderful effects, if only for one day.
Information from a site that sells this:
Alertness Without Stimulation
Adrafinil is the prototype of a new class of smart drug – the eugeroics (ie, “good arousal”) designed to promote vigilance and alertness. Developed by the French pharmaceutical company Lafon Laboratories, adrafinil (brand name, Olmifon) has been approved in many European countries for treating narcolepsy, a condition characterized by excessive daytime sleepiness and other unusual symptoms.
Non-narcoleptic users generally find that adrafinil gives them increased energy and reduces fatigue, while improving cognitive function, mental focus, concentration, and memory. It has been reported that quiet people who take adrafinil become more talkative, reserved people become more open, and passive people become more active.
Of course, many stimulant drugs, ranging from caffeine to methamphetamine, are known to produce similar alerting/energizing effects. Adrafinil has been described by some users as a “kinder, gentler” stimulant, because it provides these benefits but usually with much less of the anxiety, agitation, insomnia, associated with conventional stimulants.
Adrafinil’s effects are more subtle than those of the stimulants you may be used to, building over a period of days to months. They appear to be based on its ability to selectively stimulate 1-adrenergic receptors in the brain.2 These receptors normally respond to norepinephrine (noradrenaline), a neurotransmitter linked to alertness, learning, and memory. This is in contrast to conventional stimulants, which stimulate a broader spectrum of brain receptors, including those involving dopamine. Its more focused activity profile may account for adrafinil’s relative lack of adverse side effects.
There’s more info about Adrafinil on Wikipedia
It’s interesting that that snipped report that people become more talkative. I reported that in the original post, too, even though I didn’t realize that this was a possibility.
A good quote that I wish I could relate to better:
“Time is limited, so I better wake up every morning fresh and know that I have just one chance to live this particular day right, and to string my days together into a life of action and purpose.”
Lance Armstrong (1971 – )
Cyclist, seven-time Tour de France champion and cancer survivor2011 stuff starts here:
Awhile ago I went to a handbell festival. I took a bit of adrafinil on the main day to try to stay awake for the whole day. It didn’t seem to keep me as on as it did before. I can’t be used to it already. Maybe I’m just that much more tired than I was before.
 Our son lives in New York and every few years he gives us tickets to see a Broadway show. A couple years ago we took the train to NY to see Wicked. Usually my DH wants to go out and see sights while we’re there. I usually want to nap.
Our son lives in New York and every few years he gives us tickets to see a Broadway show. A couple years ago we took the train to NY to see Wicked. Usually my DH wants to go out and see sights while we’re there. I usually want to nap.
This time we got up on Saturday morning, went out for breakfast. I wanted to take in the whole day and enjoy Wicked so I took some Adrafinil. We got back to the hotel and got ready to go to a museum or other point of interest.
But, DH wanted to rest a bit first. Then our son closed his eyes for a bit…
So, I found myself the only one awake for the afternoon. They both work up in time for the show…
Sigh It was a great show, though.
A recent Christmas I was going to get my son some Adrafinil as a gift. The original place we bought it didn’t have any more stock so I tracked it down as a surprise. He was going to give me some, as well, but couldn’t get it from the original source, either. So he found something very similar called Modafinil. GMTA!
And 2016..
Saturday, 4/23/16 really was one of the best days I’ve had in a long time.
I’ll be writing a longer post about that later on my travel blog but here’s the original plan: https://maryoblog.com/2016/04/23/busy-saturday/
Suffice it to say, we arrived at the Tattoo and I got no nap at all, all day!


-
1
-
-


This is a tough one. Sometimes I’m in “why me” mode. Why Cushing’s? Why cancer? Unfortunately, there’s not a thing I can do about either. Cushing’s, who knows the risk factors? For kidney cancer, I found out the risk factors and nearly none apply to me. So why? But why not? No particular reason why I should be exempt from anything.
Since there’s nothing to be done with the exception of trying to do things that could harm my remaining kidney, I have to try to make the best of things. This is my life. It could be better but it could be way worse.
One of the Challenge topics was to write about “My Dream Day” so here’s mine…
I’d wake up on my own – no snooze alarms – at about 8 am, sun streaming through the window. I’d we well rested and not have had any nightmares the night before. I remember my son is home for a visit but I let him sleep in for a while.
I’d get out for a bike ride or a brisk walk, come home, head for the hot tub then shower. I’d practice the piano (or recorder or Aerophone) for a bit, then go out to lunch with friends, taking Michael with me. While we’re out, the maid will come in and clean the house.
After lunch, maybe a little technology shopping/buying. Then the group of us go to one of our homes for piano duets, trios, 2-piano music.
When we get home, it’s immaculately clean and I find that the Prize Patrol has visited and left a substantial check.
I had wisely left something for dinner in the Ninja so dinner is ready. After dinner, I check online and find no urgent email, no work that needs to be done, no bills that need to be paid, no blog challenge posts to write…
I wake up from My Dream Day and realize that this is so far from real life, so I re-read The Best Day of My Life and am happy that I’m not dealing with anything worse.

-
1
-
-
Since I’m posting this on April 21, I had a built-in topic.
http://cushieblog.files.wordpress.com/2012/04/nataile-blog.jpg?w=500
The image above is from our first local meeting, here in Northern VA – note the 6 Cushing St. sign behind us. Natalie was the Cushie in the middle.
Today is the anniversary of Natalie’s death. Last month was the anniversary of Sue’s death. I wrote about Janice earlier.
It’s just not right that this disease has been known for so many years, yet doctors still drag their feet diagnosing it and getting people into remission.
Why is it that we have to suffer so much, so long, and still there are so many deaths from Cushing’s or related to Cushing’s symptoms?
I know far too many people, good people, who suffered for many years from this disease that doctors said they didn’t have. Then they died. It’s time this stopped!
Speaking of death – what a cheery blog post this is turning out to be. NOT! Unfortunately, this seems to be one of the realities of Cushing’s.
Tomorrow will be cheerier – watch for it!

-
1
-
-
And today, we talk about pink jeeps and ziplines…
How in the world did we get here in a Cushing’s Challenge? I’m sliding these in because earlier I linked (possibly!) my growth hormone use as a cause of my cancer – and I took the GH due to Cushing’s issues. Clear? LOL
http://cushieblog.files.wordpress.com/2012/04/pink-jeep.jpg?w=300&h=225&resize=300%2C225
I had found out that I had my kidney cancer on Friday, April 28, 2006 and my surgery on May 9, 2006. I was supposed to go on a Cushie Cruise to Bermuda on May 14, 2006. My surgeon said that there was no way I could go on that cruise and I could not postpone my surgery until after that cruise.

I got out of the hospital on the day that the other Cushies left for the cruise and realized that I wouldn’t have been much (ANY!) fun and I wouldn’t have had any.
An especially amusing thread from that cruise is The Adventures of Penelopee Cruise (on the Cushing’s Help message boards). Someone had brought a UFC jug and decorated her and had her pose around the ship.
The beginning text reads:
Penelopee had a lovely time on Explorer of the Seas which was a five day cruise to Bermuda. She needed something to cheer her up since her brother, Tom, went off the deep end, but that’s another story!
Penelopee wanted to take in all of the sights and sounds of this lovely vessel. Every day she needed to do at least one special thing. Being a Cushie, she didn’t have enough spoons to do too much every day.
On the first day, she went sunning on the Libido deck……she didn’t last too long, only about 10 minutes. Goodness, look at her color! Do you think maybe her ACTH is too high?
Although I missed this trip, I was feeling well enough to go to Sedona, Arizona in August, 2006. I convinced everyone that I was well enough to go off-road in a pink jeep, DH wanted to report me to my surgeon but I survived without to much pain and posed for the header image.
In 2009, I figured I have “extra years” since I survived the cancer and I wanted to do something kinda scary, yet fun. So, somehow, I decided on ziplining. Tom wouldn’t go with me but Michael would so I set this up almost as soon as we booked a Caribbean cruise to replace the Cushie Cruise to Bermuda.
Each person had a harness around their legs with attached pulleys and carabiners. Women had them on their chests as well. In addition, we had leather construction gloves and hard hats.
We climbed to the top of the first platform and were given brief instructions and off we went. Because of the heavy gloves, I couldn’t get any pictures. I had thought that they would take some of us on the hardest line to sell to us later but they didn’t. They also didn’t have cave pictures or T-Shirts. What a missed opportunity!
This was so cool, so much fun. I thought I might be afraid at first but I wasn’t. I just followed instructions and went.
Sometimes they told us to break. We did that with the right hand, which was always on the upper cable.
After the second line, I must have braked too soon because I stopped before I got to the platform. Michael was headed toward me. The guide on the end of the platform wanted me to do some hand over hand maneuver but I couldn’t figure out what he was saying so he came and got me by wrapping his legs around me and pulling me to the platform.
After that, no more problems with braking!
The next platform was very high – over 70 feet in the air – and the climb up was difficult. It was very hot and the rocks were very uneven. I don’t know that I would have gotten to the next platform if Michael hadn’t cheered me on all the way.
We zipped down the next six lines up to 250-feet between platforms and 85-feet high in the trees, at canopy level. It seemed like it was all over too soon.
But, I did it! No fear, just fun.
Enough of adventures – fun ones like these, and scary ones like transsphenoidal surgery and radical nephrectomy!

-
1
-
-

In case you haven’t guessed it, my cause seems to be Cushing’s Awareness. I never really decided to devote a good portion of my life to Cushing’s, it just fell into my lap, so to speak – or my laptop.
I had been going along, raising my son, keeping the home fires burning, trying to forget all about Cushing’s. My surgery had been a success, I was in remission, some of the symptoms were still with me but they were more of an annoyance than anything.
I started being a little active online, especially on AOL. At this time, I started going through real-menopause, not the fake one I had gone through with Cushing’s. Surprisingly, AOL had a group for Cushing’s people but it wasn’t very active.
What was active, though, was a group called Power Surge (as in I’m not having a hot flash, I’m having a Power Surge). I became more and more active in that group, helping out where I could, posting a few links here and there.
Around this time I decided to go back to college to get a degree in computer programming but I also wanted a basic website for my piano studio. I filled out a form on Power Surge to request a quote for building one. I was very surprised when Power Surge founder/webmaster Alice (AKA Dearest) called me. I was so nervous. I’m not a good phone person under the best of circumstances and here she was, calling me!
I had to go to my computer class but I said I’d call when I got back. Alice showed me how to do some basic web stuff and I was off. As these things go, the O’Connor Music Studio page grew and grew… And so did the friendship between Alice and me. Alice turned out to be the sister I never had, most likely better than any sister I could have had.
In July of 2000, Alice and I were wondering why there weren’t many support groups online (OR off!) for Cushing’s. This thought percolated through my mind for a few hours and I realized that maybe this was my calling. Maybe I should be the one to start a network of support for other “Cushies” to help them empower themselves.
I wanted to educate others about the awful disease that took doctors years of my life to diagnose and treat – even after I gave them the information to diagnose me. I didn’t want anyone else to suffer for years like I did. I wanted doctors to pay more attention to Cushing’s disease.
The first website (http://www.cushings-help.com) went “live” July 21, 2000. It was just a single page of information. The message boards began September 30, 2000 with a simple message board which then led to a larger one, and a larger. Today, in 2022, we have over 73 thousand members. Some “rare disease”!
This was on the intro page of Cushing’s Help until 2013…
I would like to give abundant thanks Alice Lotto Stamm, founder of Power Surge, premier site for midlife women, for giving me the idea to start this site, encouraging me to learn HTML and web design, giving us the use of our first spiffy chatroom, as well as giving me the confidence that I could do this. Alice has helped so many women with Power Surge. I hope that I can emulate her to a smaller degree with this site.
Thanks so much for all your help and support, Alice!
In August 2013 my friend died. In typical fashion, I started another website…
I look around the house and see things that remind me of Alice. Gifts, print outs, silly stuff, memories, the entire AOL message boards on floppy disks…
Alice, I love you and will miss you always…
http://powersurgedotco.files.wordpress.com/2013/10/maryoonerose.gif?w=1000&resize=191%2C256
-
1
-
-

Cushie Crusader, that’s me…and many others. I think we all have an opportunity to be Cushie Crusaders every time we tell others about our illness, share our story on or offline, post about our struggles – and triumphs – on the message boards, write blog posts in this Cushing’s Awareness Challenge…
When we have prayer time in my handbell practice or choir rehearsals I try to mention issues that are going on in the Cushing’s community. People are slowly but steadily learning about Cushing’s week by week.
A piano student mentioned that a person in a group she is in has Cushing’s, a non-Cushie friend mentioned last week that she had gone with a friend of hers to an endo appointment to discuss Cushing’s.
Get out there and talk about Cushing’s. Let people know that it’s not just for dogs and horses (and sometimes ferrets)!
Here’s something I had made for Sue with SuperSue embroidered on the back.
Picture your name instead:
http://cushieblog.files.wordpress.com/2012/04/supersue.jpg?resize=425%2C600











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The Genomic Landscape of Corticotroph Tumors: From Silent Adenomas to ACTH-Secreting Carcinomas
in News Items and Research
Posted
Abstract
1. Introduction
2. Results
2.1. Clinical and Demographic Characteristics of the Patients
2.2. General Genomic Characteristics of Neoplasms of Corticotrophic Lineage
2.3. ACTH-Secreting Carcinoma (Tumor 1)
2.4. Crooke Cell Adenoma (Tumor 2)
2.5. Silent Corticotroph Adenomas (Tumors 3–5)
2.6. ACTH-Secreting Adenomas (Cushing Disease) (Tumors 6–9)
2.7. ACTH-Secreting Adenoma Causing Nelson Syndrome (Tumor 10)
2.8. Tumor Phylogenic Analysis
2.9. Correlation between Gene Variants and Clinicopathological Features
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Patients and Tumor Tissue Samples
5.2. Construction and Sequencing of Whole Exome Libraries
5.3. Bioinformatics Analysis
5.4. Sanger Sequencing forConfirmation of Exome Findings
5.5. Hormone and Transcription Factor Immunohistochemistry
5.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
From https://www.mdpi.com/1422-0067/23/9/4861/htm